
New! MegaPulse Electrophoresis Power Supply

We are pleased to introduce the MegaPulse™ (MGP0200), a new pulsed-field electrophoresis power supply that supports both Field-Inversion Gel Electrophoresis (FIGE) and Zero-Integrated-Field Electrophoresis (ZIFE) while running one or two standard agarose midi gel boxes simultaneously. With seven pre-validated protocols covering 0.5 kb to 1.6 Mb, the MegaPulse provides complete HMW DNA analysis in a single, compact instrument.
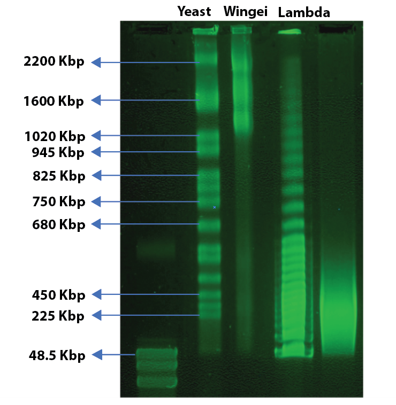
In a typical FIGE workflow, DNA fragments above ~450 kb compress into an unresolved band. ZIFE overcomes this by simultaneously switching the field magnitude, direction, and pulse time so that the net time-integrated field over each pulse cycle is near zero. Under these conditions, very large DNA molecules are resolved on the basis of the time required to reorient in response to field changes — extending practical resolution to 1.6 Mb on a standard agarose slab gel.
The MegaPulse includes seven pre-validated protocols covering 0.5 kb to 1.6 Mb across both FIGE and ZIFE modes. Users who want to go beyond the pre-sets can build and save custom waveforms using the Advanced Programming interface.
The MegaPulse is controlled by any Windows PC running our MegaPulse software. For labs that want a turnkey setup, we offer an optional mini PC tablet with touchscreen with the MegaPulse software pre-loaded. The instrument can also drive one or two Sage Pulse Midi Gel Boxes (PGB1000) simultaneously using the same protocol.
The MegaPulse is well-suited for HMW genomic DNA QC ahead of long-read sequencing library prep and any pulsed-field electrophoresis application in the 0.5 kb–1.6 Mb range.
To order, visit our product page to view available configurations and bundles. Contact us at info@sagescience.com or visit sagescience.com for more information and to download the operations manual.
New BluePippin 2.5% Agarose Cassettes Provide High Size Selection Accuracy Between 100-600bp
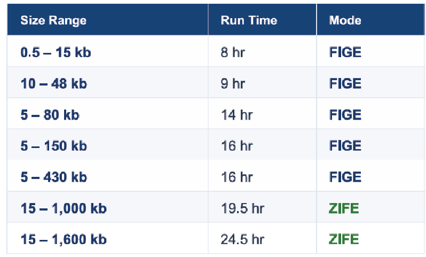
For Illumina and short-read sequencing applications, BluePippin users typically use either high-resolution 3% agarose gel cassettes and/or slightly lower resolution 2% agarose gel cassettes. The 3% cassettes are great for “threading the needle” when separating microRNA libraries from nearby unincorporated adapter dimers. The 2% cassettes provide a wider range of collection capabilities (100-600bp vs 100-250bp) with sufficient resolution for most other short-read methods.
However, we have received some customer requests for higher accuracy size selection above 250bp. So we’ve assembled a 2.5% agarose cassette to split the difference in resolution. With this cassette, we can provide better size selection accuracy and reproducibility for size selections across the same 100-600bp range.
If you feel that you can benefit from this, order part #BDF2510. These do require about a 15-25% longer run time than the 2% cassettes.
Check out the performance specification comparison between the two products below:
2.5% Agarose Gel Cassettes (BDF2510)

2% Agarose Gel Cassettes (BDF2010)
New PippinHT Cassette: >10kB High-Pass with Internal Standards
Improved Workflow for HiFi Sequencing
The PippinHT was designed to run two SBS-compatible gel cassettes, each with a 12-sample capacity, and with onboard sample tracking capability. In this way, DNA size selection could more closely match sample prep workflows undertaken on 96-well microplates. However, high molecular weight DNA size selection for single-molecule sequencing has not been optimal in this regard because HMW size selections require an external marker for calibration. This effectively reduces the sample capacity of PippinHT cassettes from 12 samples to 10 for a maximum capacity of 20 samples per run.
Since PacBio recommends the PippinHT for size selection of its HiFI WGS libraries, we developed a cassette kit that will use internal standards to calibrate size selection at the recommended 10kb threshold. This cassette will increase the capacity of the HiFi size selections to the intended 12 samples per cassette, and hopefully better compliment workflows. As an extra bonus, users can expect slightly better accuracy that is achieved with internal markers as well. The Femtopulse and Pippin Pulse gel images below illustrate similar performance to the current external marker method.

If the need arises, we can produce internal standard cassettes with higher Hi-Pass thresholds as well. So if you are running HiFi WGS, or have any application that will benefit from >10kb Hi-Pass, give this cassette a try!
Here is the ordering information: HIJ-7510, 10/pk
Decoding Wild Rice Genomes: A New Era for Climate-Resilient Agriculture
Asian rice, domesticated around 10,000 years ago, is a dietary staple for more than one third of the global population. Yet, intensive breeding for improved yield has reduced its genetic diversity – making modern varieties increasingly vulnerable to stressors like heat, drought, and salinity. In contrast, wild rice relatives have adapted to a wide range of environmental conditions providing untapped genetic material for rice crop improvement.
A landmark study, published in Nature Genetics on April 28, 2025, traced the evolutionary history of wild rice species, classified in the genus Oryza, defining their phylogenetic relationships and deciphering how their genomes have been shaped by 15 million years of evolutionary history. The research was led by Prof. Rod Wing, Director of the Arizona Genomics Institute (AGI), and his postdoctoral research associate, Alice Fornasiero, of the King Abdullah University of Science and Technology (KAUST) in Saudi Arabia, in collaboration with Wageningen University & Research in the Netherlands.
Thanks to the sequencing of both genomic DNA and RNA of 11 wild rice species (i.e. two diploids and nine tetraploids) using the PacBio Sequel II and IIe instruments, the team could successfully generate ultra-high quality genome assemblies and use accurate full-length transcripts for gene annotation. The analysis of this valuable and publicly available genomic resource allowed the construction of the Oryza pangenome – i.e. the genomic representation of the entire genus – and the description of a “core” portion remarkably stable and a “dispensable” portion mainly composed of transposable elements. Due to their ability to move and reshuffle the genome, these elements has played a crucial role in shaping the genetic diversity in these species.
The analysis of gene expression in Oryza coarctata, a species adapted to saline conditions of coastal regions, showed an unusual balance between its two subgenomes, with both contributing equally to gene activity. This equilibrium may help explain the resistance of O. coarctata to salty environments.
These findings build a genetic foundation that could help scientists breed more resilient rice varieties, or even domesticate wild species through approaches like neodomestication to generate climate-ready crops for a more sustainable agriculture.
Use of Sage technology at the Arizona Genomics Institute’s Service Center:
For the wild rice genome project described above, the genomes were sequenced in both CLR and CCS/HiFi modes. The WGS libraries were size selected with the BluePippin, using 0.75% agarose cassettes with marker U1 (for CLR libraries) or marker S1 (for CCS/HiFi libraries ).
For current projects at AGI, the team has switched from the BluePippin to the PippinHT for its size selection needs. AGI has been an early access test site for the PippinHT Range+T program mode (for improving accuracy of HMW size selection) and the soon-to-be-released PippinHT 0.75% agarose cassette internal standards (which increase sample capacity).
“Range+T “ for Tight Sizing of HMW Libraries
Range+T Size Selection
Note: Range + T is available for all PippinHT instruments. However, only BluePippins with serial numbers 2700 and above will run the method.
Sage’s DNA size selection technology has been used to help improve PacBio sequencing, particularly for mammalian and complex genome work since the introduction of the BluePippin (2012). Over the last few years, PacBio®’s Hi-Fi circular consensus sequencing has been the method that has set the gold standard for whole genome sequencing. In terms of library size, this technique benefits greatly from a sharp size cut-off at the LMW end of the size distribution (~10kb) and a slight tail at the HMW end. It also benefits from a narrow fragment size distribution around 17-18kb. A common way to accomplish this is to shear DNA with Diagenode’s Megaruptor®3 device, followed by size selection on a PippinHT or BluePippin.
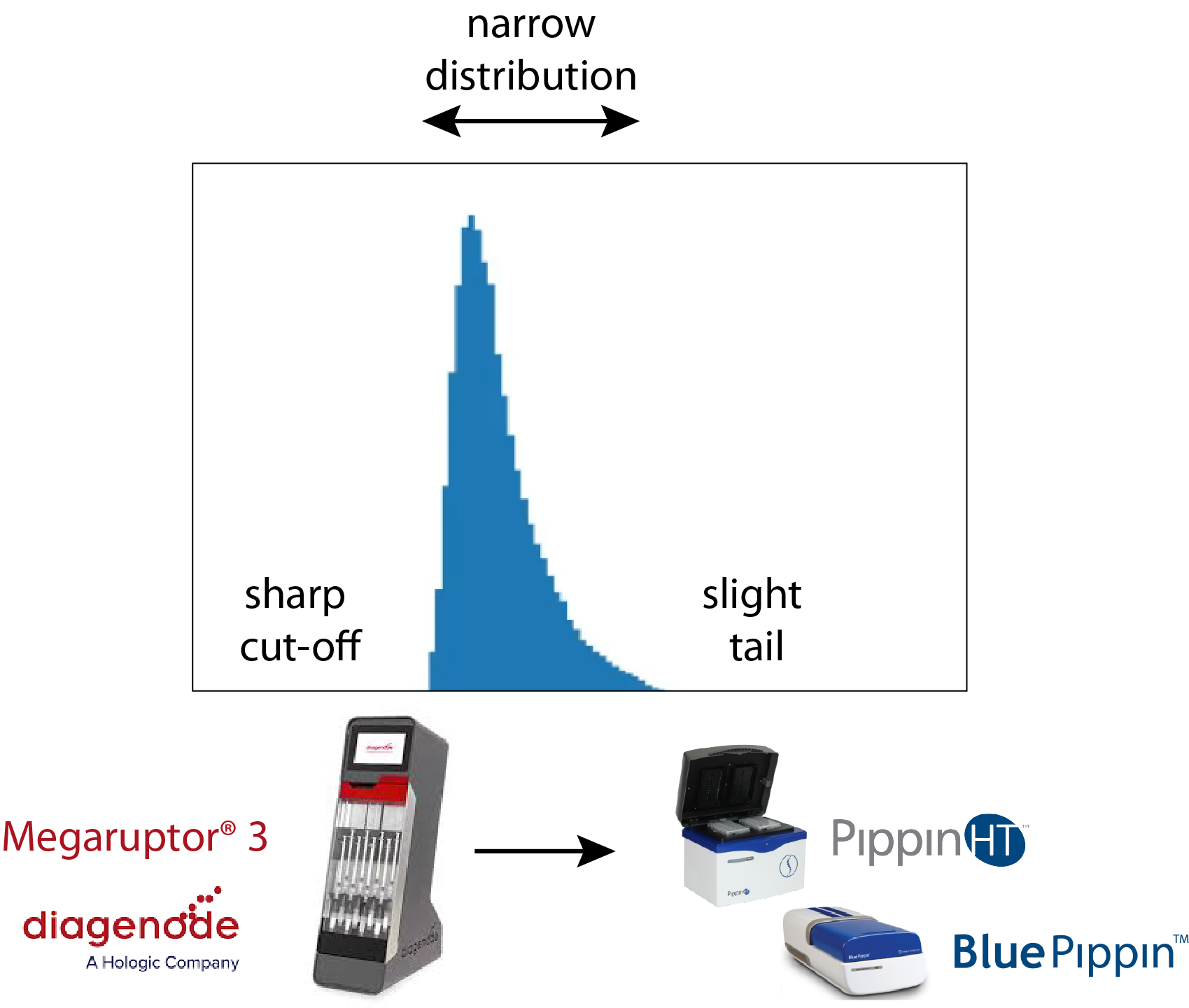
To get the desired fragment distribution at these size ranges is not trivial. In Sage gel cassettes, the total input load – and the tight fragment distribution profile of Megaruptor 3 sheared samples – affects the mobility of the DNA in a manner that cannot be easily accounted for with our standard “Range” programming method. Essentially, the start of collection of DNA is accurate and straightforward, while the timing for the end of collection can be affected by the physical nature of the sheared DNA sample. To this end, we developed a new size selection method called “Range+T”. With this method, the base pair start of collection is entered in the software and the end of collection is determined by entering a time for elution. Through a little trial and error, a time setting can be determined to overcome calibration uncertainty for the end of collection. We conducted an early access program with the Copetti lab at the Arizona Genomics Institute, resulting in increased PacBio HiFi read lengths, that you can read about in this application note.
A Collaboration with Diagenode
We decided to do a deep dive into Range+T to get a better handle the method, and to develop best practices for using Diagenode Megaruptor 3 and PippinHT or BluePippin. Working with Diagenode, we were able to process many DNA samples and experiment with multiple settings. We outlined our findings in a new application note, Best practices: Sage Science size selection with Diagenode Megaruptor shearing for long-read sequencing library preparation . We encourage you to take a look if you are interested in working with these size ranges. We found that careful coordination of Megaruptor shearing and Sage size-selection conditions are critical:
1. Megaruptor shearing conditions should ideally be chosen to provide a sheared DNA input with an average size at, or slightly above, the desired library size.
2. For accurate LMW cut-offs, start of collection DNA size on PippinHT or BluePippin should be less than or equal to the peak size (mode) of the sheared DNA input.
3. The upper boundary of the size distribution will be determined by the Megaruptor shear conditions if long Sage Range+T elution times are used (i.e., 30 minutes or longer).
4. Sage Range+T programming can provide significantly narrower size distributions by shortening the Range+T elution time below 30 minutes.
For size selection, provided the same Megarupter shear conditions (and input amount) are used, we recommend starting with Time values at 5 minute intervals (ie. 5,10,15,20 minutes) to determine the best distribution conditions. It is very important to enter a starting base pair value that is before the mode of the Megaruptor distribution – so QC your samples!

Range+T is available now for both PippinHT and BluePippin. It requires a software upgrade (v.6.41 and v1.14, respectively, available for download on sagescience/support). We have also packaged cassette kits specifically for this purpose; HRT7510 and BRT7510. Starting Range values can be between 9-30kb.