AGBT23 Roundup!
Well, we have another fabulous AGBT behind us that we were happy to attend. Our take home messages? A. Spatial transcriptomics is the exciting new frontier. B. True human WGS has been accomplished and is moving into population-scale work. And C. There are a lot of new sequencing technologies being bandied about.
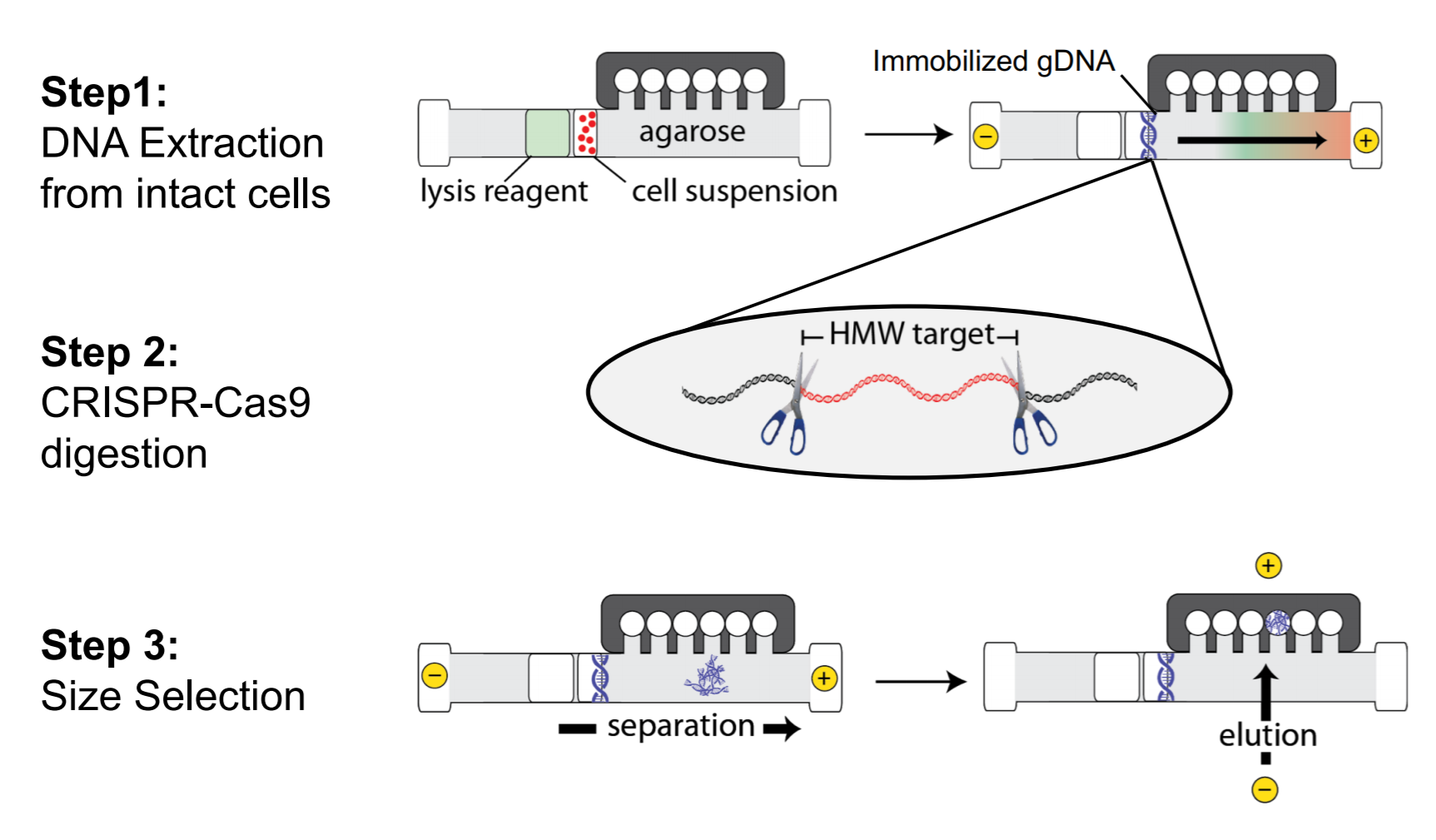
Though we weren’t presenting or introducing, Sage Science did have a few key mentions. The Broad Institute described their efforts to automate a PacBio HiFi whole genomes sequencing pipeline for the NIH’s All of Us program. In their poster, you can view it here, they go into quite a bit of detail why gel size selection (via the PippinHT) is the preferable method with HiFi library construction.
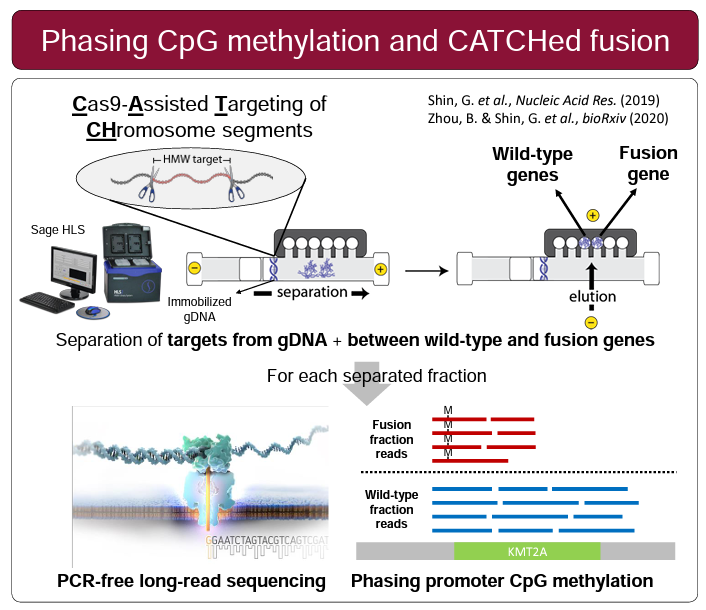
HLS-CATCH, our method for enriching high molecular weight targets made an appearance in a few posters. Universal Sequencing, our frequent collaborators and partners (we also sell their TELL-seq kits) presented a nice demonstration of their linked-read technology. They show haplotype phasing data for larger targets (200kb with HLS-CATCH) and smaller targets (4-20kb) using PCR and andCas9 Exo/Pulldown method (a modified CaBagE protocol).
Three posters were presented that featured HLS-CATCH and Oxford Nanopore sequencing. Researchers from the Ji lab at Stanford University presented a very compelling poster on KMT2A translocations, a common chromosomal abnormality in leukemias. Using HLS-CATCH (they use the term CLTR-Seq) and nanopore sequencing they identify rearrangement structure as well as the methylation landscape. Stanford researchers from the Ji and Urban labs presented a poster on a study in which HLS-CATCH is used to sequence segmental duplications, also by nanopore sequencing, associated with the neuropsychiatric 22q11.2 deletion region. Finally, CATCH was used in a study presented by Gavin Arno from the University College London and collaborators that is focused on inherited retinal disease and complex rearrangements associated with the OPN1LW/OPN1MW gene array. The authors, like the Stanford researchers, conclude that CATCH-nanopore sequencing is effective unraveling genomic mysteries that are “intractable to NGS”.
HLS-CATCH and TELL-Seq: Haplotype Phasing of a 187kb HMW Gene Targets from a Trio
We here at Sage Science are excited to have joined forces with Universal Sequencing Technology to promote their TELL-Seq barcode linked-read technology. One of the reasons for our excitement is that TELL-Seq, and the transposase-based barcoding method benefits greatly from high molecular weight DNA – one of Sage’s areas of expertise. Our own HLS-CATCH method is one area that is a particularly nice fit with TELL-Seq particularly given the relatively small amount of DNA that HMW target purification yields. The easy library workflow and ready access to Illumina sequencing is also a big plus.
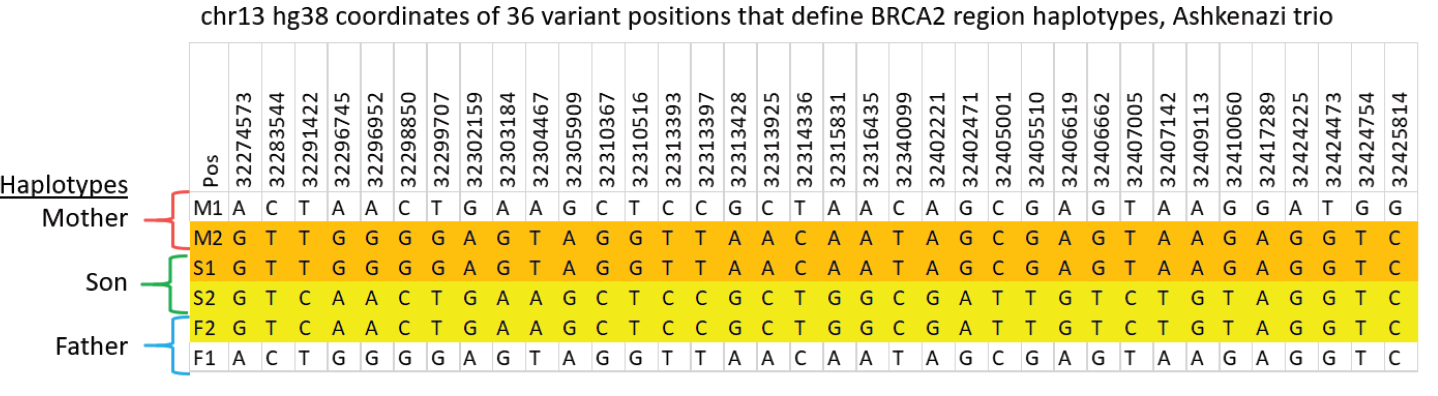
Validating this proposition was simple and straightforward. We performed HLS-CATCH using cultured cells from the Genome in a Bottle Ashkenazi trio sample set using a Cas9 guides that we designed to purify a 187kb target containing the BRCA2 locus. TELL-seq libraries were prepared from the targets from each individual, sequenced and run through 10X Genomic’s Long Ranger analysis package and then visualized the data on the 10X Loupe browser. The phase blocks sizes were 186 kb (mother), 181kb ( son), and 168Kb ( father), and only a small-genome scale analysis was required. Download our whitepaper here.

The variants in the 6 haplotypes compared favorably to the Genome in a Bottle high quality sequences.
Clearly CATCH and TELL-seq can be an economical alternative for obtaining long range genomic data for gene targets. The value of this technique was previously demonstrated in a Stanford University paper (Shin, GW, et al. 2019) where, among other analyses, the 4Mb MHC histocompatibility complex was phased using the 10X Genomics linked read method. The simplicity of TELL-Seq is great news vis a vis the effort required for long-read sequencing libraries. However, strides are being made there as well. Two recent papers have demonstrated great results using HLS-CATCH with PacBio sequencing (Walsh, T. et al. 2020) and Oxford Nanopore (Zhou, B, et al. 2020).
Unleash Your Illumina Sequencer with TELL-Seq™ Linked Reads
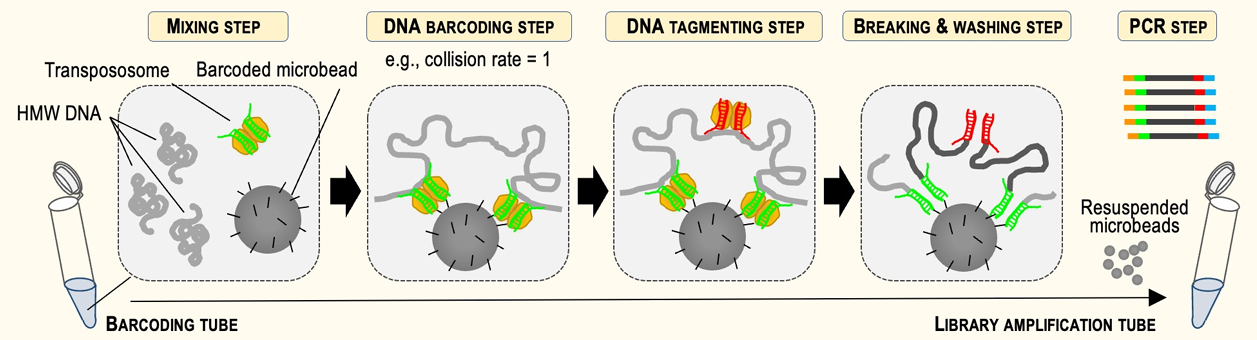
We’re proud to announce today that we will be selling TELL-Seq™ linked library prep kits from Universal Sequencing Technology. TELL-Seq, Transposase Enzyme Linked Long-read Sequencing, is a scalable NGS library technology with a very simple workflow that can provide you with long-read information with your illumina® sequencer. This can all be done in a single tube, with as little as 0.5 – 5 ng of genomic DNA. The higher the molecular weight of the genomic DNA, the better, the transposase does the rest. Check out the applications here and the technology here, and some FAQs here.
Watch the video below to see how TELL-seq works:
TELL-Seq and TELL-Bead are trademarks of the Universal Sequencing Technology (Canton, MA)
HLS-CATCH and TELL-Seq™: a new route to Targeted Long-Fragment Linked-Read Sequencing
At the recent 2020 AGBT meeting Universal Sequencing gave a data-filled poster on their new Transposase Enzyme Linked Long-read Sequencing kit and workflow, known as TELL-Seq™. The technology features a unique combination of bead-linked and soluble transposases that can generate linked-read libraries in 3 hours from as little as 0.1ng of genomic DNA input. Their kits offer all of the advantages of linked-read sequencing without the need for microfluidic instrumentation. The scientific poster can be viewed here.
One of the featured projects was a collaboration with Sage Science combining Sage’s HLS-CATCH targeted long-fragment sample prep with TELL-seq. Sage prepared a 200kb genomic DNA target from the BRCA1 locus (from an anonymous blood donor) on its SageHLS platform and sent the product to Universal Sequencing labs for TELL-seq sequencing. The data clearly revealed both haplotypes over the entire 200kb target region. In addition, de novo assembly of the BRCA1 reads revealed a small 1.5kb heterozygous deletion in the 5’ regulatory region of the gene.
The data demonstrated that the combination of TELL-seq and HLS-CATCH will be a powerful, high-resolution, and cost-effective option for long-range genome phasing and targeted de novo sequencing and assembly.
An example of a phased assembly from the HLS-CATCH/TELL-Seq workflow (Tom Chen, et al. presented at AGBT 2020)

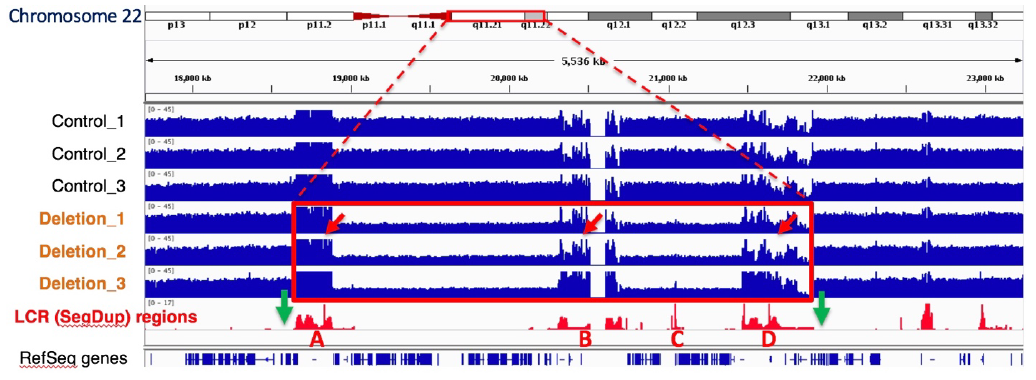
SageHLS Helps Unravel the Neuropsychiatric 22q11.2 Deletion Region1q11
An international collaboration led by the Stanford labs of Alexander Urban and Hanlee Ji, used the SageHLS system to map a Mb-sized deletion in a complex region of the human genome, 22q11.2, that is associated with a variety of neurodevelopmental and neuropsychiatric disorders. Structural variation (SV) in this region is difficult to study because the breakpoints are frequently located in four large segmental duplications (up to 400kb in length) that are scattered over a 3 Mb area. Conventional short-read sequencing cannot unambiguously identify the breakpoints of these SVs because of the high homology between the segmental duplications. The researchers designed an HLS-CATCH procedure in which Cas9 was used to cleave unique sites outside of the 3 Mb region that contains the segmental duplications. In a patient sample carrying a large deletion, a ~300kb CATCH product from the deletion allele could be cleanly separated from the much larger 3 Mb CATCH product from the wild-type allele by preparative pulsed-field electrophoresis in the SageHLS. The researchers were then able to sequence the 300kb deletion product using Oxford Nanopore long-read methods and successfully map the deletion breakpoints.
{kind=link}
Some of this work was presented in a scientific poster at the recent AGBT 2020 conference at Marco Island. The poster can be viewed here.
The green arrows below indicate the Cas9/RNA target sites that flank the 22q11.2 region (Bo Zhou, et al. presented at AGBT 2020)