Broad Institute Teams with Sage Science for Automated Sizing
Don’t miss this great blog post from the Broad Institute (“A Sage partnership”) describing collaborative work between their genome sequencing team and Sage Science to design a better size selection process for the Broad’s sequencing pipeline.
Headed up by Sheila Fisher, assistant director of technology development for the Broad’s Genome Sequencing Platform, the goal was to replace error-prone, tedious manual gel extractions in the sample prep workflow. Working with Sage’s Pippin platform, Sheila and her team were able to automate the size selection step, improving accuracy and eliminating the chance for cross-sample contamination.
An added bonus was that Pippin sizing offered much higher yields than manual gel extraction had, allowing Fisher’s team to accept samples with just 100 nanograms of DNA, instead of the 3 or 4 micrograms the pipeline originally required. “This opened up a significant number of samples to the process that we couldn’t sequence before,” says Sheila in the blog post. “We were able to build a very strong partnership with Sage, and the result was a true co-development project.”
We couldn’t have said it better ourselves. It’s truly a pleasure to continue with our great collaboration with the Broadies!
Automated Size Selection Key for RADseq: PLoS One Publication
Congratulations to Sage Science customer Hopi Hoekstra and her team at Harvard University for their recent publication in PLoS One! Dr. Hoekstra, who works in the Organismic & Evolutionary Biology and the Molecular & Cellular Biology departments at Harvard, reports a full laboratory protocol for RADseq, or reduced-representation genome sequencing for use in population genotyping. The method allows for studying hundreds of thousands of markers across hundreds of individuals or more.
The paper, published on May 31 and entitled “Double Digest RADseq: An Inexpensive Method for De NovoSNP Discovery and Genotyping in Model and Non-Model Species,” can be found here. (link to paper)
The authors write: “Our method requires no prior genomic knowledge and achieves per-site and per-individual costs below that of current SNP chip technology, while requiring similar hands-on time investment, comparable amounts of input DNA, and downstream analysis times on the order of hours.”
As part of the library prep protocol, the Harvard team tested out manual gel extraction versus the Pippin Prep for size selection. The paper reports that manual gel excision did not perform as well as automated size selection, “likely because [gel excision] was imprecise or ‘leaky.’” The authors note that for manual gel electrophoresis, “careful practitioners can achieve roughly 50% of the precision and repeatability of automated DNA size selection.”
For more on the Hoekstra lab, click here. (link to her lab)
At Emory, Size Selection Saves Time for NGS Prep
At the genome center at Emory University, scientists credit the Pippin Prep with shaving almost a full day off the sample prep process for Illumina’s mate-pair library prep.
Chad Haase, laboratory manager at the Emory GRA Genome Center, says that the Sage Science Pippin Prep size selection instrument his lab acquired about a year ago has replaced the 16-hour runs that had previously been done on gels with a very low agarose concentration.
Jamie Davis, a scientist at the genome center, says that she typically runs the Pippin “after we do our first end repair and the biotinylation reaction.” Size selection generally takes about an hour, she says.
The Pippin Prep also works well for 454 sequencing. Haase’s team had tried another automated gel system, but it wouldn’t work for fragments larger than 1 kb. “Once we started making the large mate-pair libraries — 3 kb, 8 kb, and 20 kb fragments — then we had to come up with something different and the Pippin system was the best,” he says.
“With the large kb mate-pair libraries, the Pippin has really been our savior there,” Haase says. “A sample prep process that used to take us three days, we can now get down to roughly two days.”
Haase says that his team also uses the Pippin before amplicon sequencing on the Roche/454 GS-FLX Titanium. “If you have any small fragments whatsoever in a 454 amplicon library, it’s going to ruin the run,” he says. “The emulsion PCR prior to sequencing is preferential to smaller fragment amplification, so you’d get a whole bunch of 50-base-pair sequencing reads” without using the Pippin for size selection.
New Cassettes Use Internal Standards
We’ve just released new dye-free cassettes for the BluePippin and Pippin Prep that use dye-labeled DNA markers* as internal standards, instead of typical external marker set. In the new product, the labeled markers will come premixed wi
th the Ficoll loading solution, and will be mixed and loaded with the sample DNA in each lane. The internal standards are designed to run well ahead of sample DNA targeted for elution, so that little or no marker contamination of eluted samples should occur.
These new cassettes will offer the following benefits:
- Faster run times. Internal standards automatically correct for the lane-to-lane mobility variation that occurs at higher voltage. Using the new high voltage 1.5% cassettes, collection of DNA fractions as large as 500 bp can be carried out in 35 minutes or less.
- Improved accuracy and reproducibility. Compared to dye-free cassettes run with an external marker, the accuracy and reproducibility of DNA extractions** will be improved (~ 5% vs. 10%). We have found the reproducibility to be quite good:
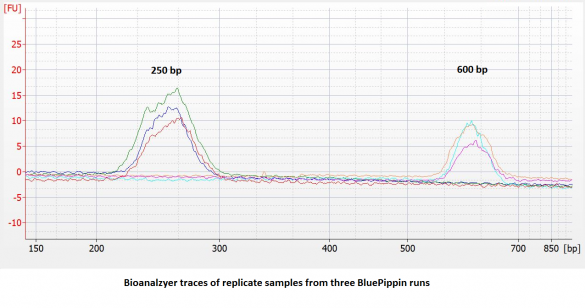
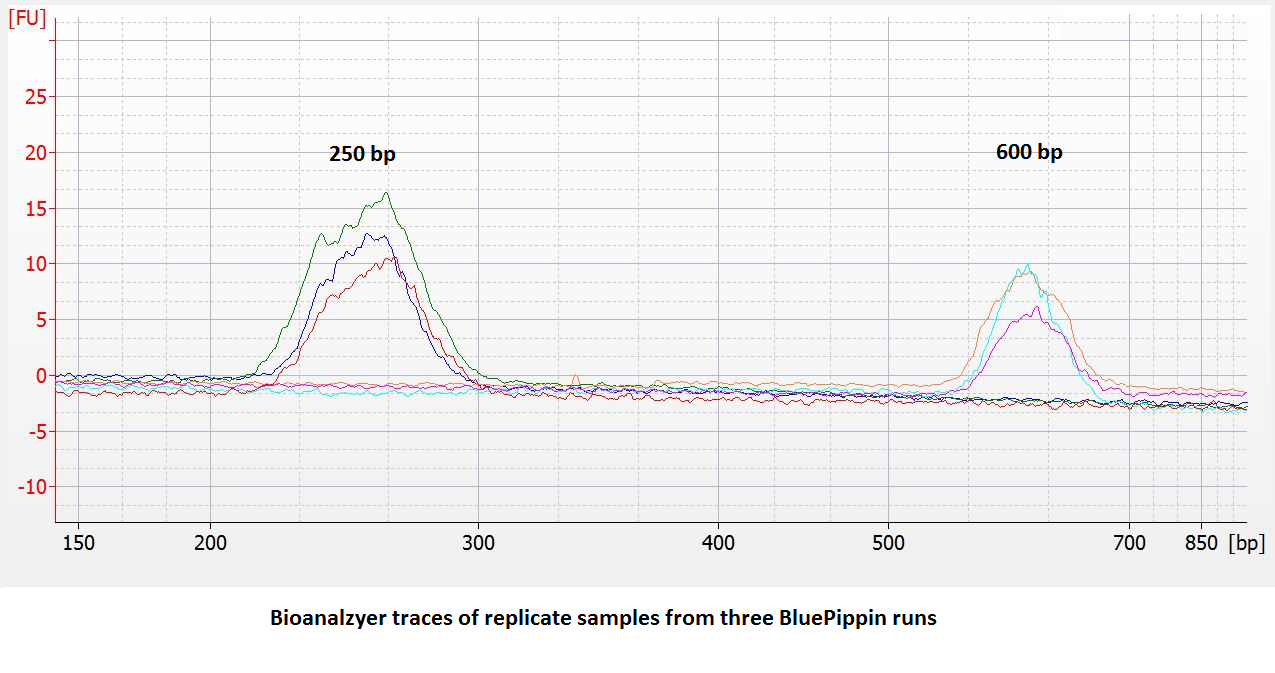
- 25% higher sample capacity. All five lanes can be used for samples. (This also means the per-sample cost goes down by 20%.)
- Use fewer lanes per run without penalty. Users may run a single sample, re-tape the open ports and save the remaining 4 lanes for later. With external standards, each run requires at least 2 lanes: one for external marker and one for sample.
Will internal standards contaminate your sequencing result?
The BluePippin internal standards are designed to run well ahead of the usable size range of the cassette. This minimizes the chance for marker contamination in eluted sample DNA since the platform excels at eliminating sample contamination from lower molecular weight fractions. In addition, when fractionating adaptered libraries, sequencing artifacts caused by marker DNA contamination are expected to be minimal, since the marker fragments are not complementary to amplification primers used by the major sequencing platforms. This expectation has been demonstrated on the Illumina platform (Tech Note in process). To enable identification/filtering of marker sequence, we’ve posted the sequences on our support page (www.sagescience.com/support). You can click the link below to get there:
http://sagescience.com/wp-content/uploads/2014/08/Internal-standard_sequences-UPDATED-082014.pdf
Input load and accuracy
The 1.5% DF cassettes can accommodate up to 10 ug of genomic input DNA. However, there are load-dependent changes in DNA mobility in these cassettes. Since most of our customers are running DNA samples of 2ug or less, we have changed our standard input load for calibration to 2 ug. Most users will find excellent accuracy for input loads up to 2 ug. At higher input loads, users should be prepared to run some preliminary tests to determine the best settings for their application. In general, at loads greater than 2 ug, selected DNA size will be slightly higher than programmed. As input load increases, so will the deviation from the programmed value.
Look or ask for these cassettes for the BluePippin:
For the BluePippin
- BDF1510 – 1.5% agarose with internal standards for the BluePippin. 250 bp-1.5 kb.
- BDF3010 – 3% agarose with internal standards for the BluePippin. 90 bp-300 bp.
For the Pippin Prep
- CDF1510 – 1.5% agarose with internal standards for the Pippin Prep. 250 bp-1.5 kb.
- CDF3010 – 3% agarose with internal standards for the Pippin Prep. 90 bp-300 bp.
* We use Mirus fluorescent label with our markers. “www.mirusbio.com”
** Accuracy = Deviation of the actual target base pair value from software input value divided by the actual value expressed as a percentage. (In calibration, actual DNA sizes are determined using an Agilent Bioanalyzer 2100.)
Reproducibility = 2X standard deviation of replicate samples expressed as a percentage of the average value.
“Overflow Detection” definition on the Pippin Prep to be eliminated
| |
Pippin Prep users are likely familiar with our “overflow detection” cassette definitions that are found in the “Cassette Type” drop down menu of the Protocol Editor. We use these definitions because Pippin cassettes exhibit a phenomenon
where the elution module liquid volume increases with prolonged sample collections*. To prevent users from inadvertently overflowing the elution modules, we embedded limits in the software to restrict users from programming broad collection ranges.
However, overflowing is remedied by sealing the elution port with tape (which we now provide with our cassettes, or any PCR tape is fine). This keeps the volume constant and prevents the elution module from overflowing. For users using taped elution modules we removed the programming limits, and created the “No Overflow Detection” cassette definition. As the cassette list becomes more extensive, we’re trying to streamline options to make things easier and more uniform for users . Here’s a screenshot of our current list:

For our next software release, we’ll simplify the definitions (see below) such that they all effectively have “No Overflow Detection” – so please be careful. We recommend using tape for all extractions. If you don’t, be careful, or use the “Tight” programming mode.

*Overflowing is due to an electro-osmotic effect caused by the properties of the ultrafiltration membrane in the elution module.