Pippin Pulse
Field Inversion Gel Electrophoresis

We’ve set up this post to provide Pippin Pulse users with a resource to keep updated on new protocols and to post references that might be useful. We also invite users to feel use this page as a forum, if you see fit, and we’d enjoy showing interesting gel images and data (email these to support@sagescience.com).
Our go-to book on theory:
“Electrophoresis of Large DNA Molecules:Theory and Applications” (E. Lai and B. Birren, eds), Current Communication in Cell & Molecular Biology Vol. 1,. Cold Spring Harbor Laboratory Press, New York, 1990
Download the Pippin Pulse user manual here
Our Gel Set-up
We use a Galileo Model 1214 RapidCastTM Mini Gel Unit running 12 X 14 cm gels using Lonza SeaKem® Gold Agarose. We use our Pippin Tris-TAPS buffers (which you can order directly from us in the US and Canada, Part No. KBB1001, under “Accessories” on our ordering page) or 0.5X TBE. The formulations can be found in the Pippin Pulse user manual or separately here.
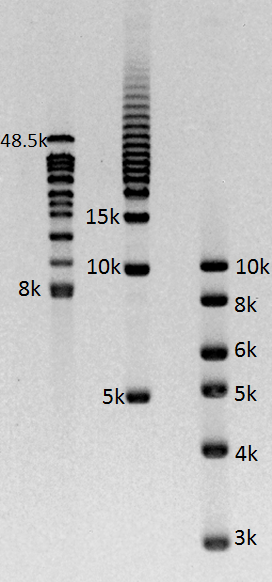
Gel images of our 1-50kb and 3-70kb protocols:
(click to enlarge images)

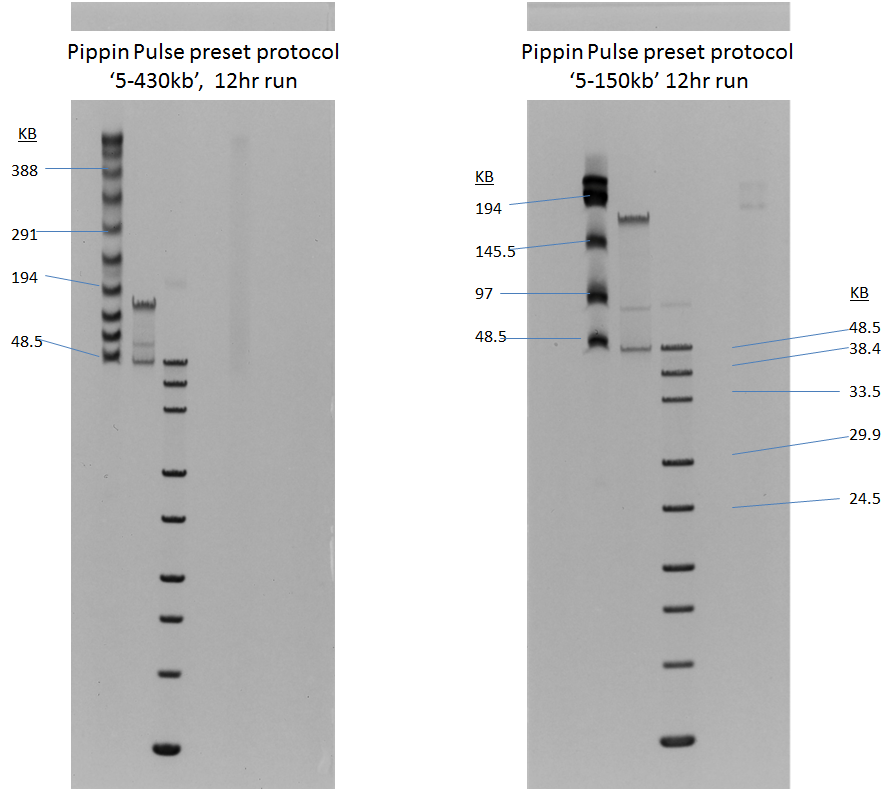
Gel images of our 5-430kb and 5-150kb protocols:
Improved Recovery for 3-10kb DNA Size Selection
BluePippin users, we have a new cassette definition that will appear in the Cassette Definition Set 10 (CD10) that we feel merits some further detail. This definition is named “0.75% DF 3 – 10kb Marker S1 – Improved Recovery”
What is it?
This definition improves the sample recovery for customers using BLF7510 Marker S1 cassette kits for DNA size selection, for size ranges between 3- 10 kb. The original cassette definition for this range, “0.75% DF 3 – 10kb Marker S1” uses pulsed-field during elution (size selection), and we have subsequently found that direct-current elution improves recovery.
Who should use it?
This is now the recommended cassette definition to use for size-selecting between 3-10kb. However, if you have optimized a protocol using the current version you may want continue to use it (it will not be deleted as a cassette definition option) if you do not wish to re-optimize.
Also note that since the BluePippin system does not allow for simultaneous pulsed-field and direct-current operation, lanes that are being separated using pulsed-field will temporarily be halted during every elution process. This makes the total run times lengths dependent on the elution times (and size selection targets), and therefore more difficult to estimate and can add up to 40 minutes to a cassette run.
How much improved is the recovery?
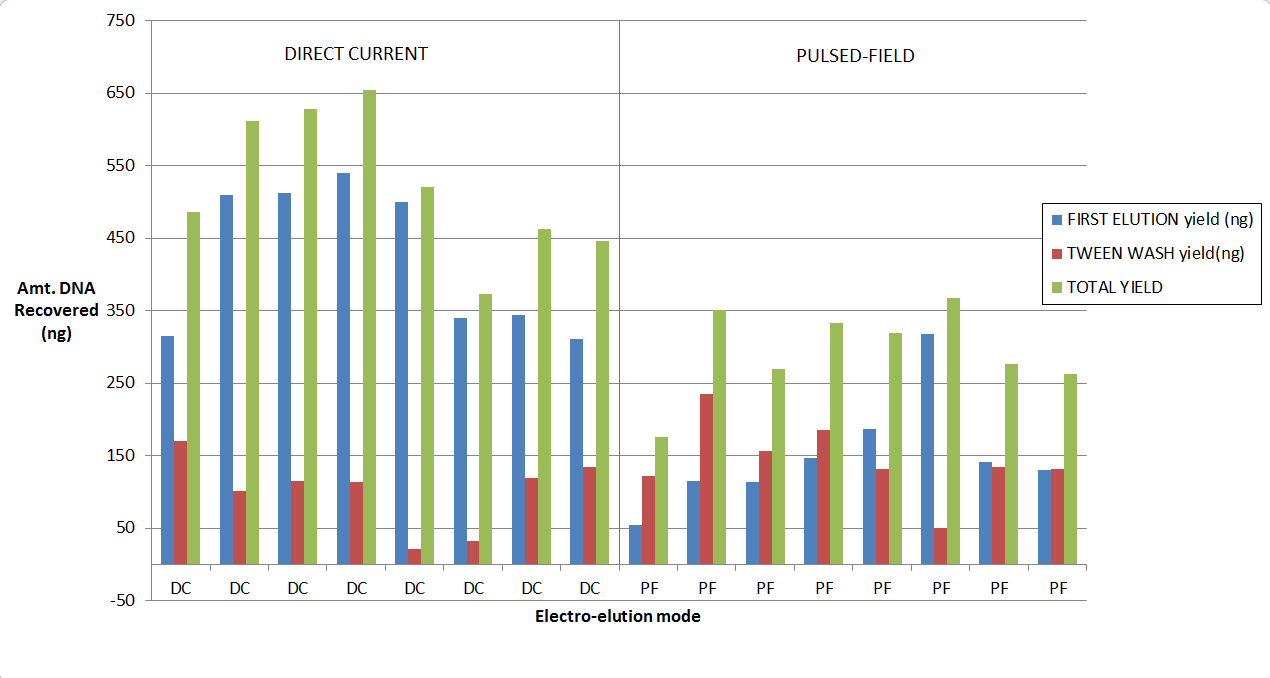
Our validation of the change in the cassette definition shows up to a 2-fold increase in recovery. As you may know, for collections of large fragments (on 0.75% agarose cassettes) we provide Tween solution which we find improves recovery if one employs a post-collection Tween rinse. Considered together, here are some results from our study. We started with a 5 ug of restriction digested DNA, and 8kb “Tight” collections:
At TGAC, Pippin-Aided Sequencing Gives Quality Boost to Genome Assemblies
Before a new technology is deployed at The Genome Analysis Centre in Norwich, UK, it must run the gauntlet of Matthew Clark’s lab.
Clark is the sequencing technology development leader at TGAC, where part of his role involves testing out new platforms, deciding whether they’d be a good fit at the institute, and ironing out best-practice workflows for the ones that are chosen.
The BluePippin automated size selection platform from Sage Science is one of the tools that succeeded in Clark’s technology proving ground and is now used more broadly throughout TGAC, where major projects include sequencing the wheat genome and studying honeybees. Clark, who joined the institute in 2010 after spending seven years as a scientist at the Wellcome Trust Sanger Institute, says that TGAC researchers rely on Pippin for building long-insert libraries for sequencing projects.
These projects include de novo sequencing, for which precise size selection in mate-pair libraries offers a significant improvement in quality of the genome assembly. The mate-pair sequence might represent a small fraction of the total data generated for the assembly — most will come from the shorter-insert paired-end libraries — “but it has a massive effect on the quality of the output,” Clark says. “The bigger-insert library gives you a 5x or 10x jump in quality, maybe even bigger, in terms of the sizes of the assembly that you’re able to generate.” These libraries, which are sequenced on Illumina or Pacific Biosciences instruments, offer longer-range information that can fill gaps or jump over repeats and other problematic structures that would otherwise break up an assembly. Indeed, Clark says the TGAC bioinformatics team prefers Pippin-aided sequencing libraries because the tight size selection helps them determine how far apart certain reads should be and put together a more accurate assembly.
Clark’s team had tested other automated size selection options, but Pippin was the best platform for generating these valuable large-insert libraries. (Pippin Prep can yield libraries with inserts up to 8 kb, while BluePippin works for even longer inserts.) It also allows for loading more material than other size selection alternatives, making it a good fit for the no-PCR paired-end libraries that some TGAC scientists like to run, Clark says. “If you have enough DNA, you can just skip the PCR — and you get much better coverage across the genome and a better assembly. That’s easier to do on a Pippin.”
NHGRI Scientists Find Transgenic DNA Has More Rearrangements than Expected
Amanda DuBose didn’t set out to come up with a new technical solution to characterizing transgenic DNA in animal models. A postdoctoral fellow in Francis Collins’ lab at the National Human Genome Research Institute, DuBose was using mice to study the premature aging disease Hutchinson-Gilford Progeria Syndrome (HGPS) when she and her colleagues realized that they were hindered by their inability to determine which mice were homozygous for the transgene containing the mutated gene of interest. This was crucial information: mice that are homozygous for the transgene with the HGPS-causing variant have a more severe phenotype with an earlier onset, making them more useful for research than the heterozygotes.
The scientists tried a number of tools — including qPCR, FISH, and Southern blots — but none of the assays could quickly and reliably report the number of gene copies in a mouse. So the team resorted to what DuBose calls “an invention of necessity” and made a custom microarray to identify the site of transgene integration and develop a genotyping assay that could deliver the results they needed.
The results of that work were published in Nucleic Acids Research in a paper entitled “Use of microarray hybrid capture and next-generation sequencing to identify the anatomy of a transgene.” Lead author DuBose and her colleagues analyzed regions of the transgenic mouse genome next to BAC sequence by using arrays to capture the DNA, size selecting with the Pippin Prep, and sequencing on the Illumina HiSeq.
DuBose, Collins, and the other authors found that the BAC had broken more than expected when it was injected into the mouse; there were four mouse-human junctions. “I was surprised by how rearranged it was,” DuBose says, noting that the junctions within the BAC were quite unexpected. “We know it’s not unique to our model. I have a feeling that all of the transgenic models have crazy rearrangements — it’s just that nobody goes in to look this closely.”
Ultimately, DuBose used the sequence data to design a PCR assay with three primers crossing one of the BAC-mouse junctions. That assay is now a standard tool that can be used to determine gene copy number in the model mice: results indicate whether a mouse is homozygous mutant, heterozygous mutant, or wild type.
The lab has now returned to its HGPS research, but DuBose says that the technological approach to characterizing a transgene and developing a PCR genotyping assay can be used by anyone working on transgenic models or genome sequence that has foreign DNA in it. She notes that the array her team used for targeted capture is no longer available, so the technique would have to be adapted for liquid capture.
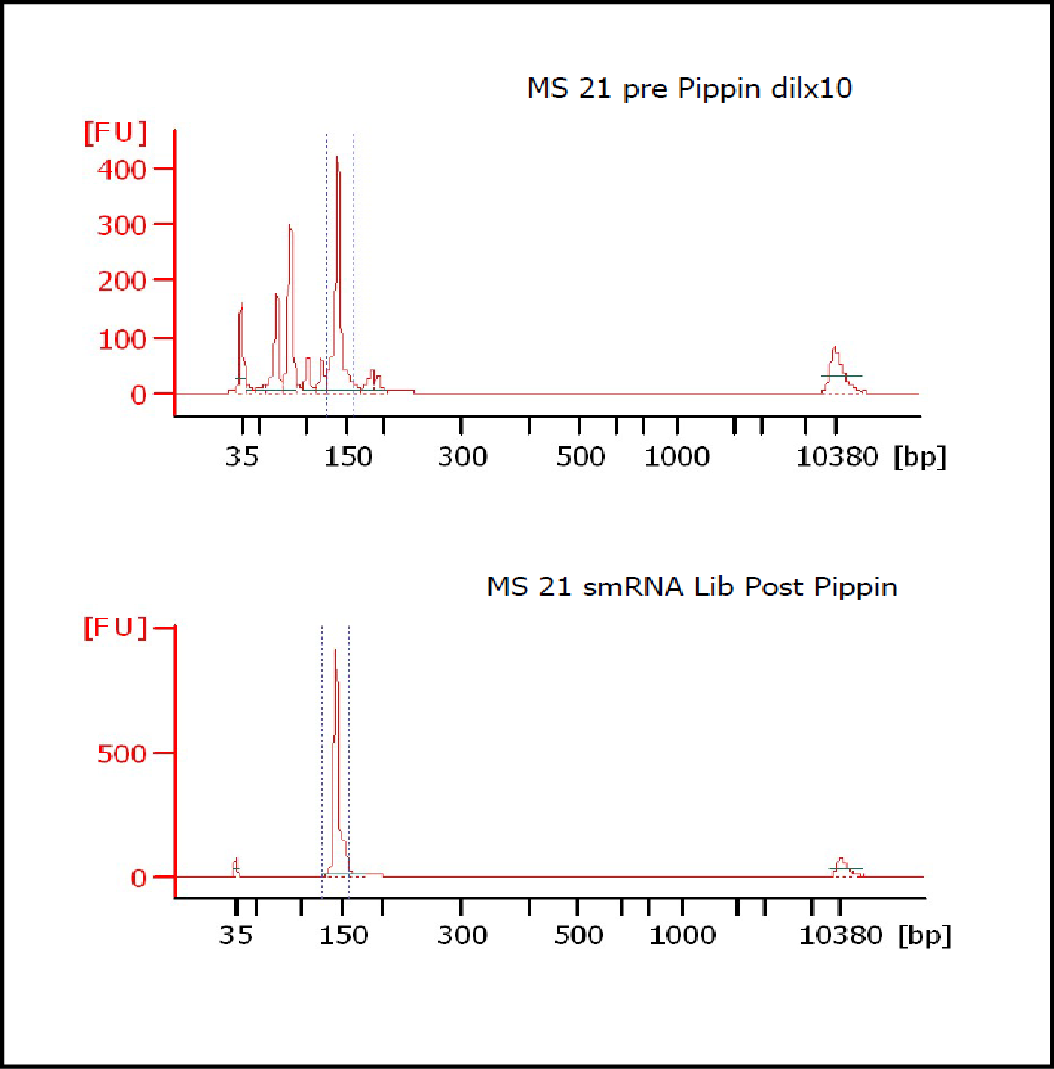
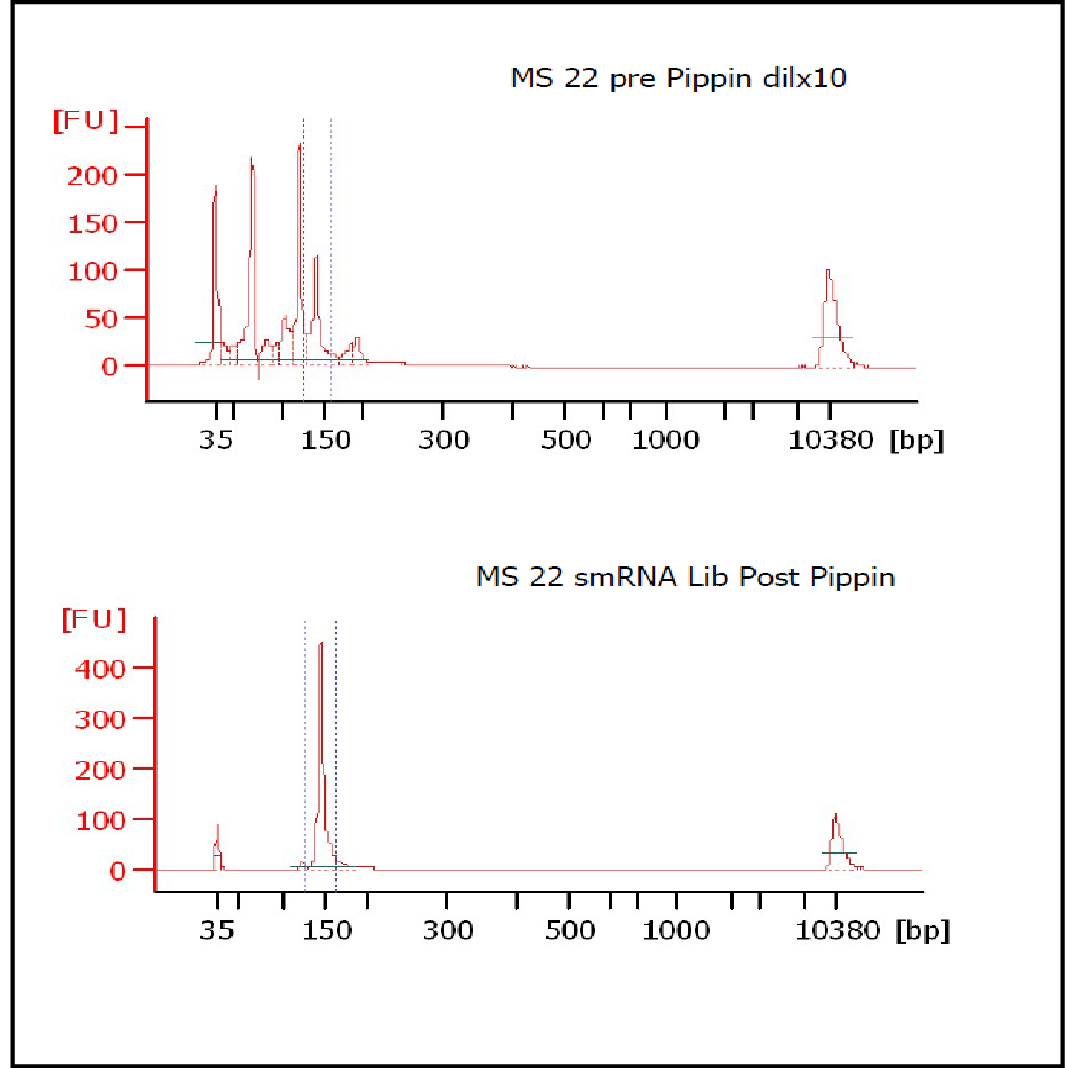
At Iowa Core Lab, Pippin Sizing Is Essential for MicroRNA Studies
Kevin Knudtson’s DNA Facility at the University of Iowa acquired a Pippin Prep almost a year ago, and in the time since, the automated size selection tool has become an integral part of the core lab’s microRNA pipeline. “Pippin is part of that workflow,” says Knudtson, director of the lab. “We’re not even going to consider a manual gel extraction for microRNAs.”
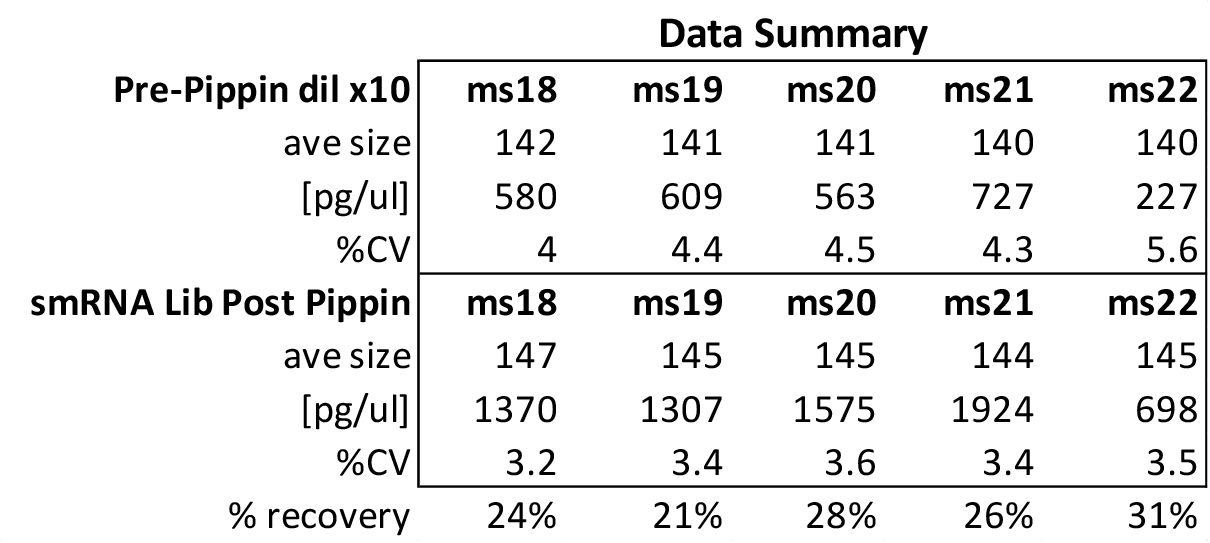
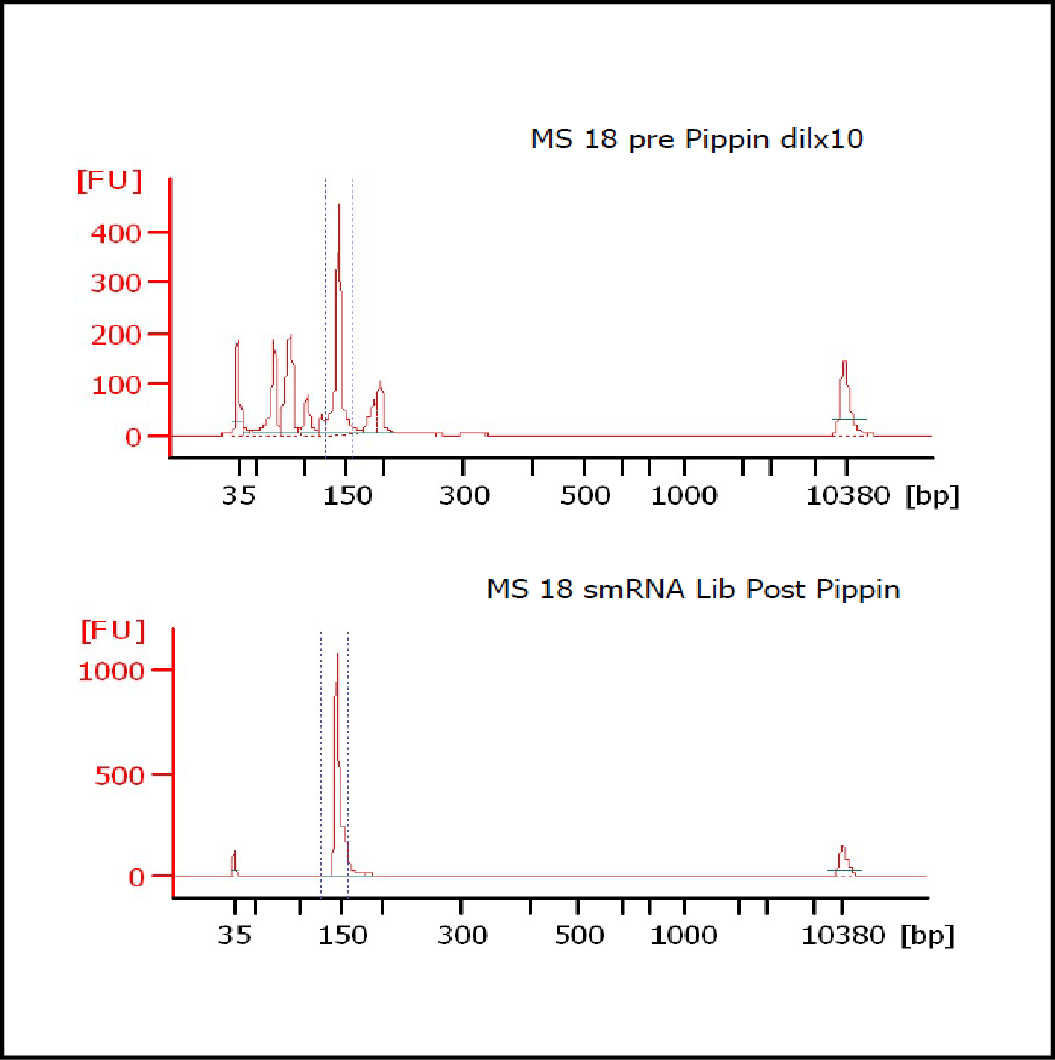
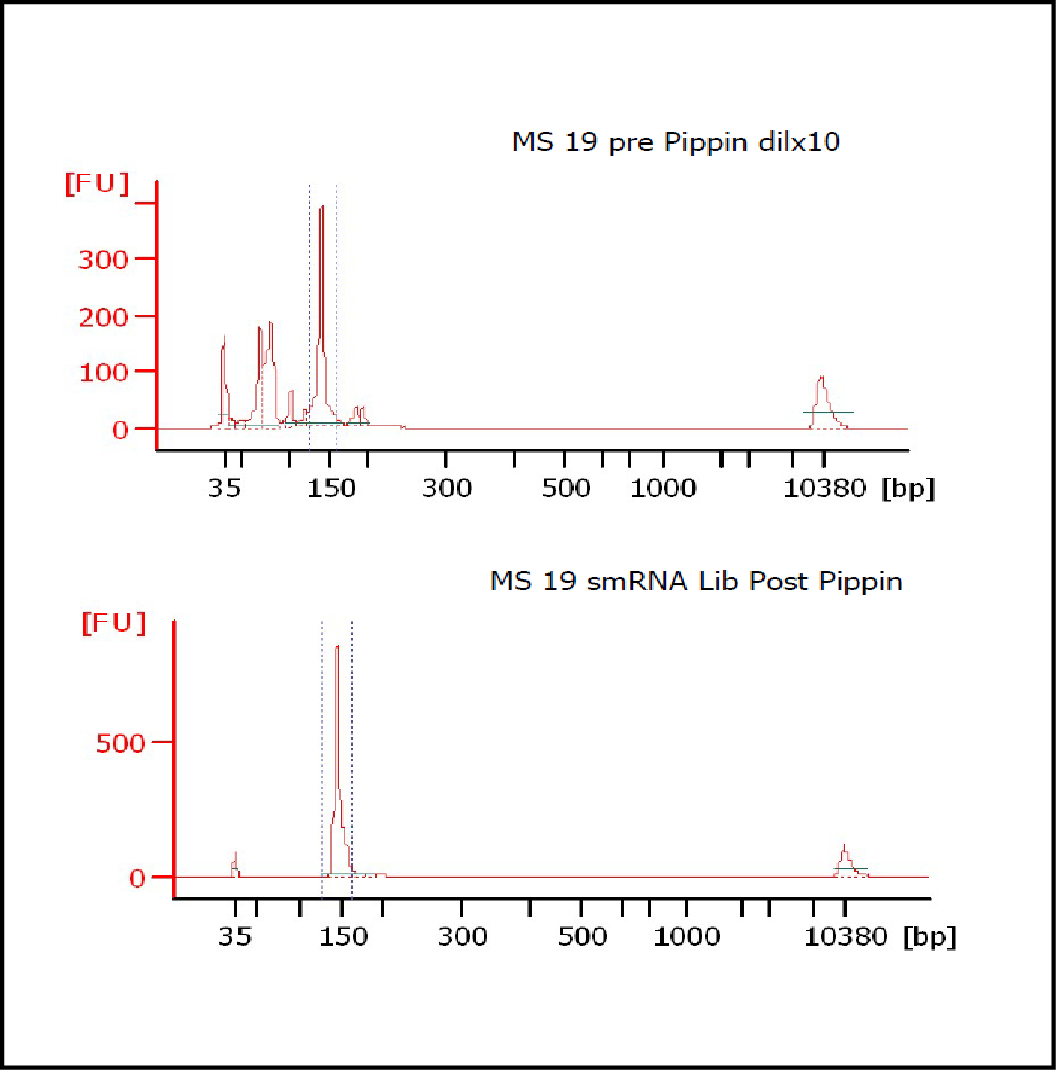
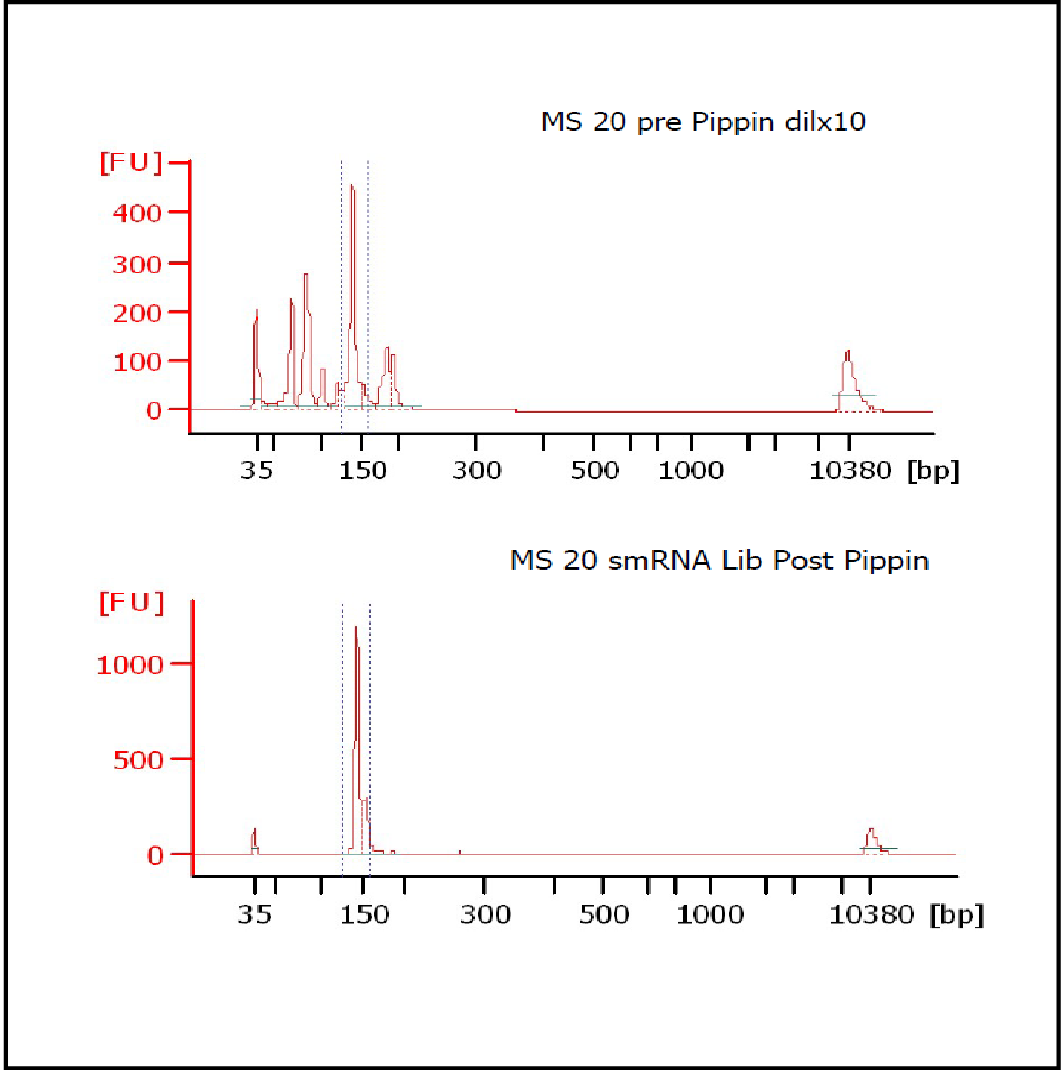
MiRNAs are especially tricky to extract from gels because the band of interest is near other bands containing unwanted products that adversely affect the quality of the sequencing run. “When you look at a Bioanalyzer trace, the band we’re after really is pretty small,” says Jennifer Bair, a member of Knudtson’s lab. “You see a lot of the other peaks much better.” The miRNA range of interest might be just 145 bases to 160 bases in size, but there will be quite a lot of adapter-dimers, primers, and other smaller content, as well as RNAs larger than the desired target. “We’re always amazed that the Pippin can isolate such a small fraction with such accuracy,” Bair adds. “It’s great.”
“For a microRNA process, there are a couple of bands that you do not want to collect,” Knudtson says. “Effectively excluding the unwanted or contaminating bands is essential to having good-quality data.”
The core facility brought in the Pippin Prep size selection instrument from Sage Science last year to replace manual gels, which are time-consuming and prone to inconsistency. “Cutting a slice out of a gel is a very subjective thing to do, and it can be technically challenging to repeat that same cut,” Knudtson says. “But when we run these out on the Pippin Prep, we’re getting exactly what we hope to receive. I can have different technicians do the same procedure and essentially get the same band or answer back when they’re processing samples.”
Knudtson’s team deploys automated sizing for more than the miRNA workflow. It has been useful for next-gen sequencing sample prep in the Iowa lab, which has a range of instruments: 454, HiSeq, MiSeq, and PGM. It takes a lot of samples to feed that pipeline, Knudtson notes. “Anything we can do to streamline the workflow is appreciated,” he says. “The Pippin Prep helps to shorten that process — it has sped up the turnaround time of samples.” Automated size selection also contributes to more efficient loading of flow cells, letting Knudtson get the most bang for his buck with sequencing runs.
The scientists have also begun to use the Pippin for ChIP-seq experiments, in which shearing chromatin to the correct size can be a real challenge, says Bair. While they are still evaluating how much size selection helps with this particular process, Bair says that generally the libraries can be made more efficiently without the larger fragments that sometimes get incorporated during the ChIP pulldown step.
Knudtson is pleased to have the days of manual gels behind him. From now on, he says, “if the protocol requires a gel extraction, we’re going to use the Pippin Prep to do it.”