At Iowa Core Lab, Pippin Sizing Is Essential for MicroRNA Studies
Kevin Knudtson’s DNA Facility at the University of Iowa acquired a Pippin Prep almost a year ago, and in the time since, the automated size selection tool has become an integral part of the core lab’s microRNA pipeline. “Pippin is part of that workflow,” says Knudtson, director of the lab. “We’re not even going to consider a manual gel extraction for microRNAs.”
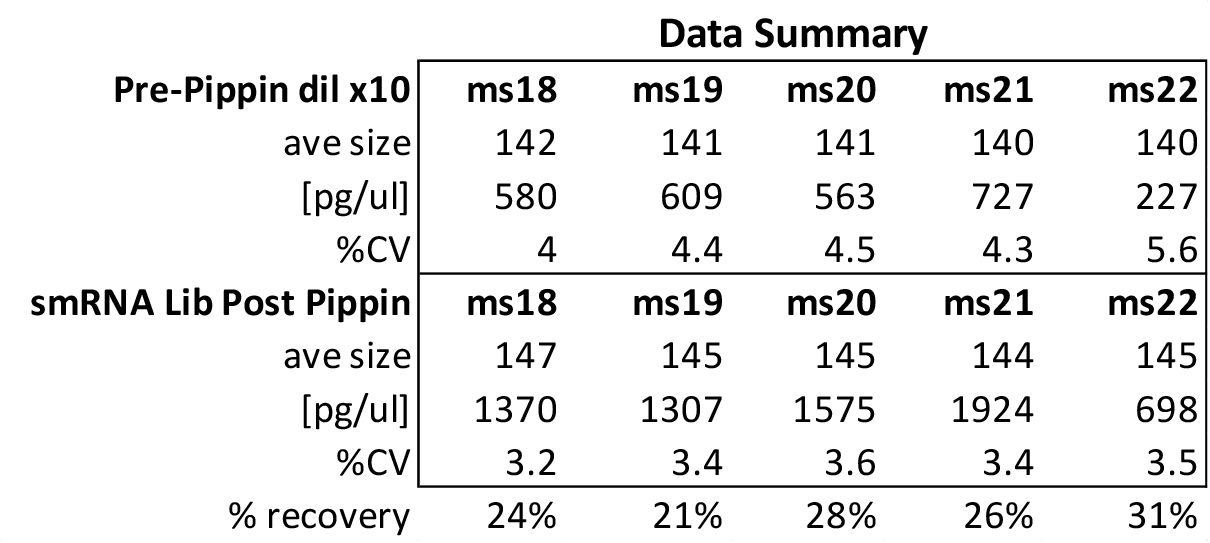
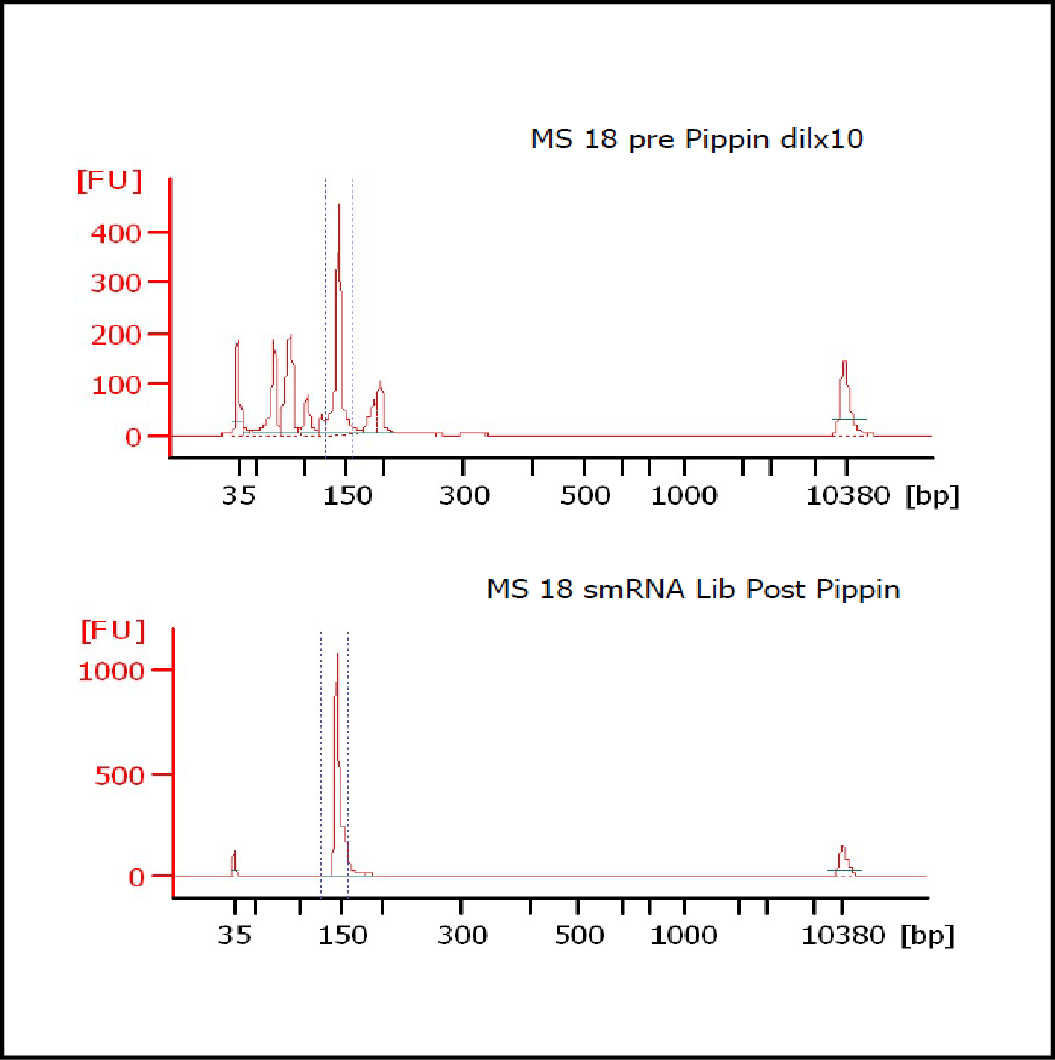
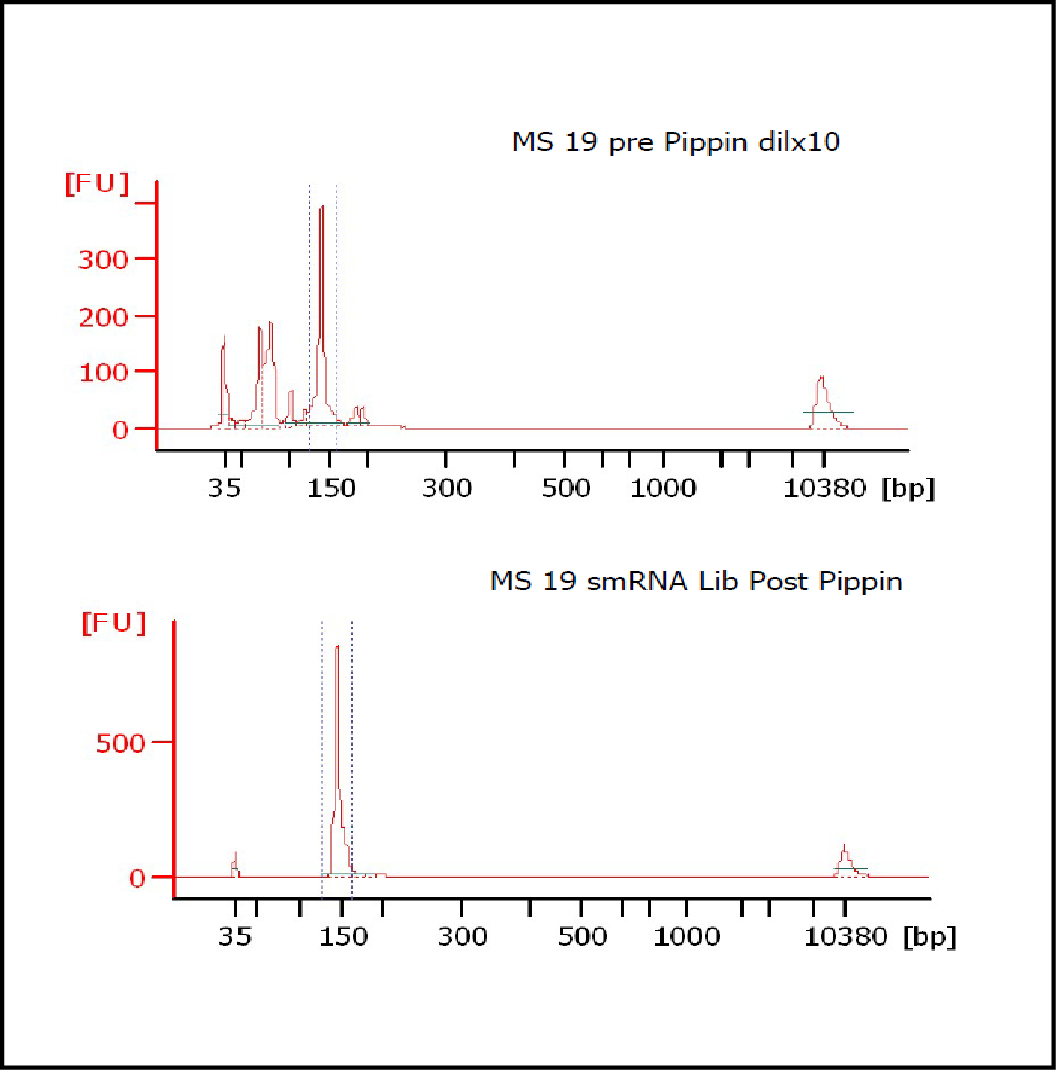
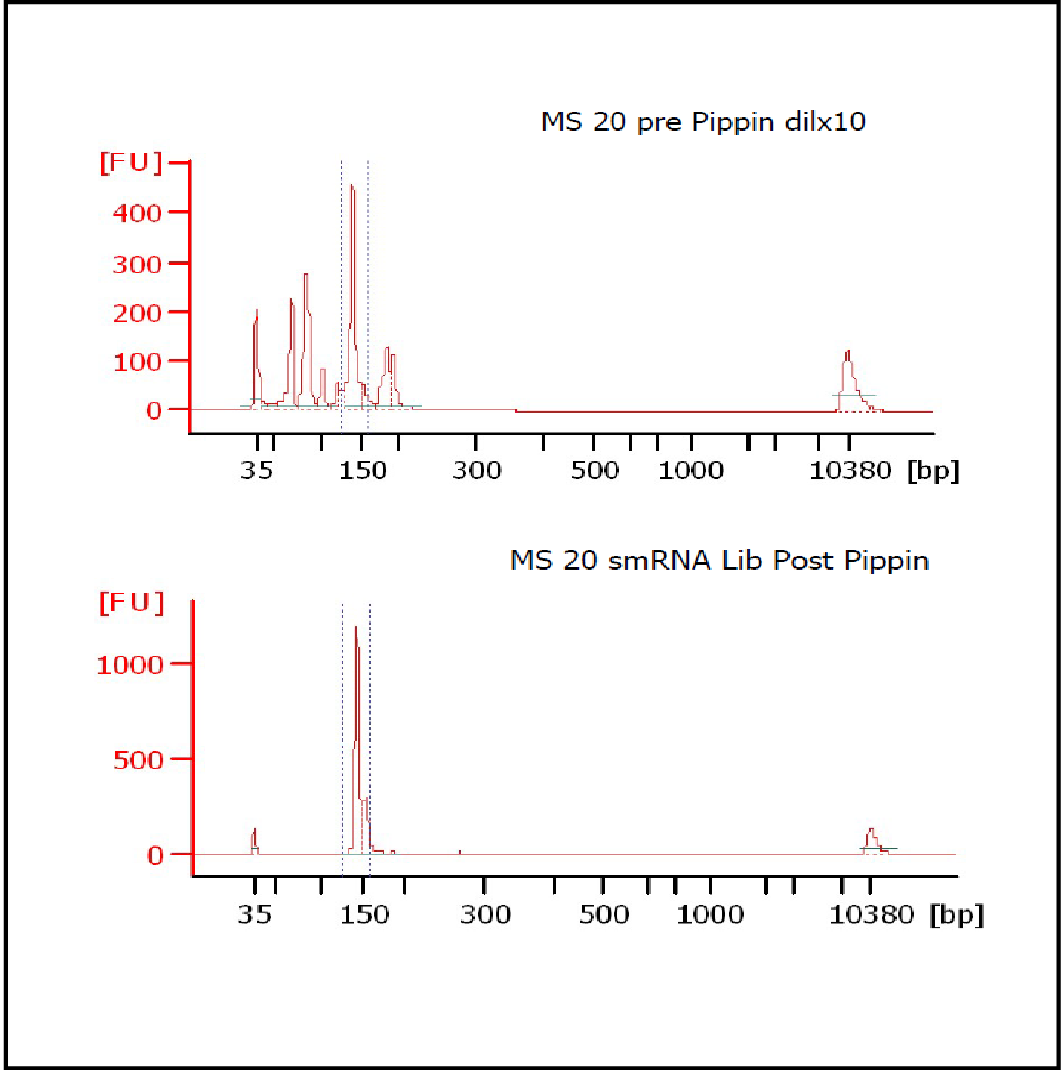
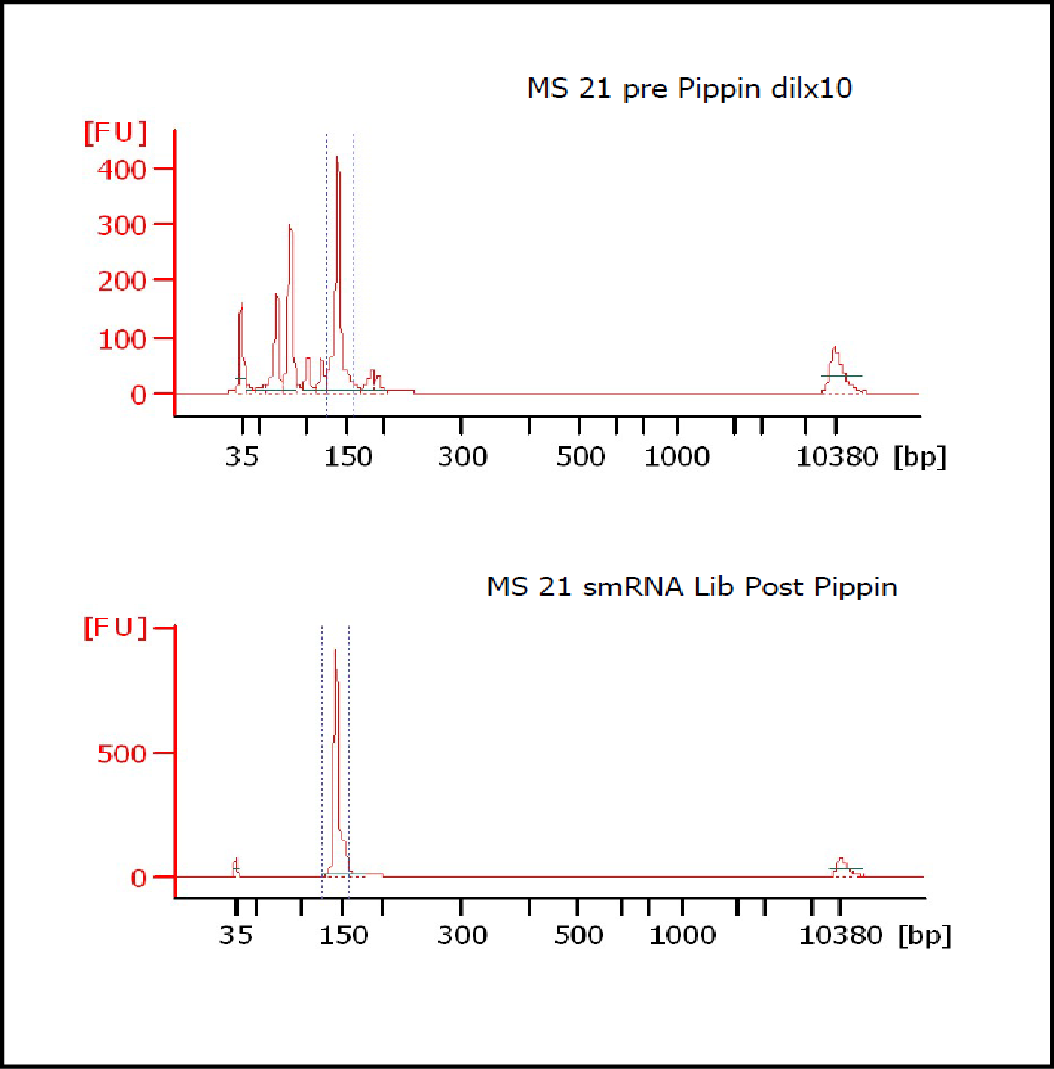
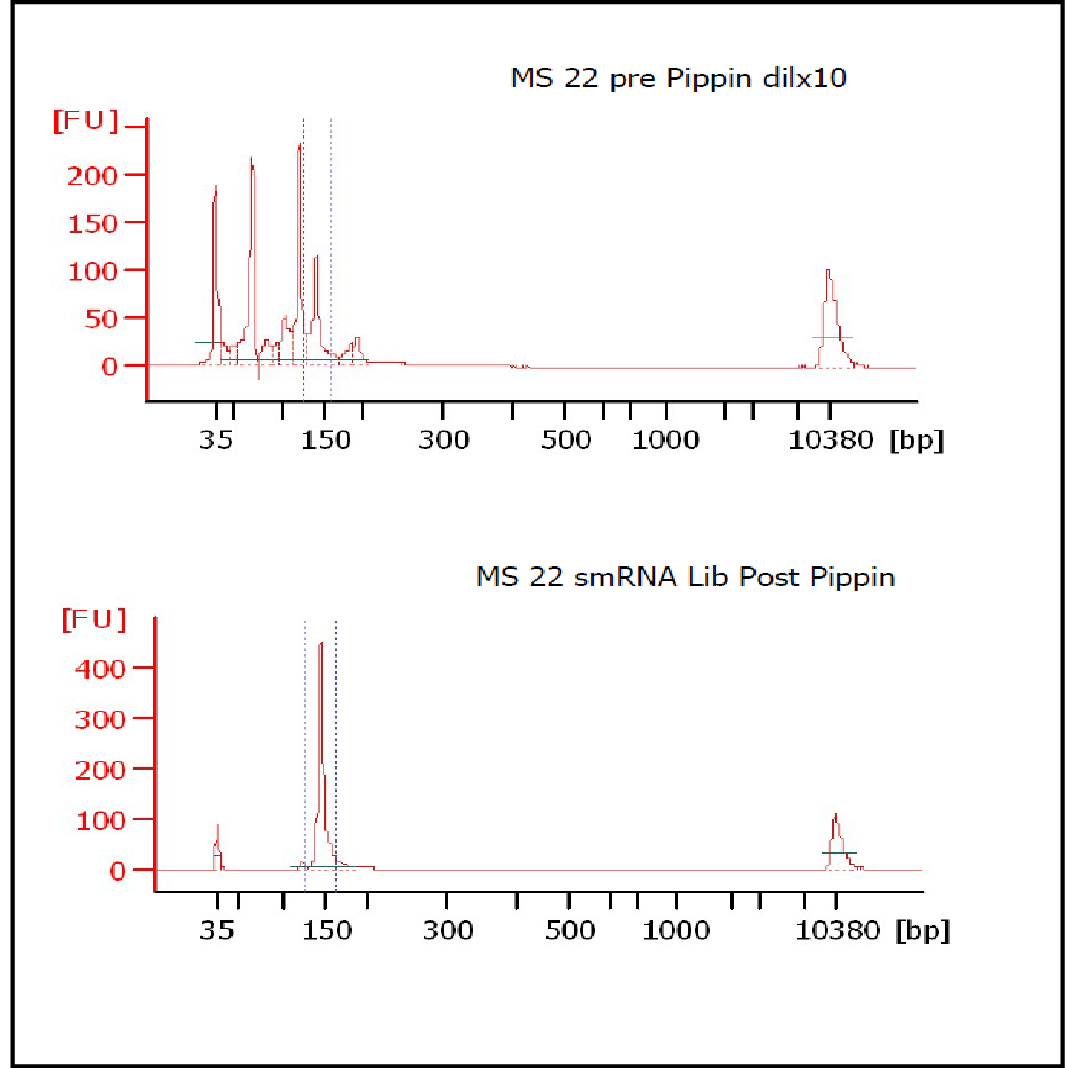
MiRNAs are especially tricky to extract from gels because the band of interest is near other bands containing unwanted products that adversely affect the quality of the sequencing run. “When you look at a Bioanalyzer trace, the band we’re after really is pretty small,” says Jennifer Bair, a member of Knudtson’s lab. “You see a lot of the other peaks much better.” The miRNA range of interest might be just 145 bases to 160 bases in size, but there will be quite a lot of adapter-dimers, primers, and other smaller content, as well as RNAs larger than the desired target. “We’re always amazed that the Pippin can isolate such a small fraction with such accuracy,” Bair adds. “It’s great.”
“For a microRNA process, there are a couple of bands that you do not want to collect,” Knudtson says. “Effectively excluding the unwanted or contaminating bands is essential to having good-quality data.”
The core facility brought in the Pippin Prep size selection instrument from Sage Science last year to replace manual gels, which are time-consuming and prone to inconsistency. “Cutting a slice out of a gel is a very subjective thing to do, and it can be technically challenging to repeat that same cut,” Knudtson says. “But when we run these out on the Pippin Prep, we’re getting exactly what we hope to receive. I can have different technicians do the same procedure and essentially get the same band or answer back when they’re processing samples.”
Knudtson’s team deploys automated sizing for more than the miRNA workflow. It has been useful for next-gen sequencing sample prep in the Iowa lab, which has a range of instruments: 454, HiSeq, MiSeq, and PGM. It takes a lot of samples to feed that pipeline, Knudtson notes. “Anything we can do to streamline the workflow is appreciated,” he says. “The Pippin Prep helps to shorten that process — it has sped up the turnaround time of samples.” Automated size selection also contributes to more efficient loading of flow cells, letting Knudtson get the most bang for his buck with sequencing runs.
The scientists have also begun to use the Pippin for ChIP-seq experiments, in which shearing chromatin to the correct size can be a real challenge, says Bair. While they are still evaluating how much size selection helps with this particular process, Bair says that generally the libraries can be made more efficiently without the larger fragments that sometimes get incorporated during the ChIP pulldown step.
Knudtson is pleased to have the days of manual gels behind him. From now on, he says, “if the protocol requires a gel extraction, we’re going to use the Pippin Prep to do it.”
Pippin Shines in Size Selection Comparison from Sanger
A paper that came out December in the journal Electrophoresis compares Pippin Prep size selection with Caliper’s Labchip XT and finds that Pippin gives “very tight size distributions” not seen with other sizing methods.
“Evaluation and optimisation of preparative semi-automated electrophoresis systems for Illumina library preparation” from lead author Michael Quail and colleagues at the Wellcome Trust Sanger Institute, demonstrates several types of comparisons between the systems. The authors note in their premise that better sizing techniques are needed since manual gels are time-intensive and lack reproducibility while SPRI beads often give ranges “too broad for de novo assembly applications.” The authors assessed size selection methods for general library preparation, PCR bias, noPCR use, and more.
The importance of accurate size selection goes beyond removal of small fragments, including adaptors and adaptor-dimers, to improve de novo sequence assembly and allow for chromosomal rearrangement prediction, the authors note.
In their first test for whether the instruments performed according to their specifications, “both platforms collected the specified size fractions but with quite different results,” Quail et al. write. “The Pippin Prep gave very tight size distributions whereas those from the Labchip XT were much broader. … The distribution of recovered fragment sizes was much more reproducible with the Pippin Prep.” Such tight sizing can lead to lower yield, the authors note, adding that “this can partially be overcome by setting the instrument to collect a wider size range.”
The study also looked at the effects of performing size selection prior to PCR amplification, finding that the tightest possible size fraction was advantageous before amplification. “Whilst Pippin Prep eluted DNA, size selected using the ‘tight’ setting, gave sharp and representative size distributions following PCR, we regularly observe smaller fragment ‘shoulders’ … following amplification of Labchip XT fractionated ligation products,” the scientists write.
While this paper focused on sample prep for the Illumina sequencing workflow, the authors note that Pippin works well with other next-gen technologies too. “We have found the Pippin Prep to be useful for both size fractionation of 100bp and 200bp fragments for the Ion Torrent PGM, and for size fractionation of 3kb fragments for Pacific Bioscience sequencing,” they write.
At MIT, Pippin Enables Splice Variant Detection and MicroRNA Analysis
At the BioMicro Center at MIT, Director Stuart Levine, PhD, recently introduced the Pippin Prep from Sage Science to enable key applications — including splice variant analysis with RNA-seq and microRNA analysis — that were not possible on other platforms.
Levine joined the BioMicro Center about four years ago; in that time, he has transformed it from a two-person lab to a major genomics and bioinformatics core facility with a dozen full-time employees serving more than 80 MIT faculty members. “My goal was to create an integrated core which would work with labs on everything from experimental design through to experimental analysis and anything they need help on anywhere along that chain,” Levine says.
His team of technology experts supports faculty members from a number of departments and institutes, covering scientific areas as diverse as cancer, environmental health sciences, biological engineering, and more. Levine’s core lab colleagues are professional scientists who focus on tools and methods, so they can translate that expertise across scientific areas and tailor experiments to each customer. “Ultimately what comes into our lab is nucleic acid, and what we do with nucleic acid is relatively constant,” Levine says. “The technology improvements that are useful for, say, understanding the evolution of ocean ecosystems can also apply to cancer research.”
In the last few years, Levine added sequencing sample preparation steps, and says that he realized early on he would have to find alternatives to manual gels. As a chargeback facility, Levine has to base prices on the fully-loaded costs of any service. When it came to setting prices for manual gel procedures, “I had to budget how long it takes to pour a gel, run out the sample — one per gel to avoid contamination — cut out the band, isolate the band. And when I put a price tag on it, I know that price tag is high enough that absolutely no one will pay for it. Once you start adding in the labor costs, the economics don’t make any sense at all,” he says.
Levine has been using the SPRIworks System from Beckman Coulter Genomics, which has been very successful as a gel alternative. However, certain applications require tighter sizing than was possible with that system, which is why he decided to look at the Pippin platform.
“What Pippin let us do was get into areas that we hadn’t been able to before,” Levine says. Two of those areas were RNA-seq — particularly identifying splice variants — and miRNAs, he adds. On the miRNA front, his team would otherwise have had to use manual gels and cut out bands, but “we wouldn’t be able to do that with any kind of economy of scale,” he says. “The high percentage gels on the Pippin allow us to cut out bands of the right size for microRNAs.”
When it comes to splice variant analysis, Levine says that his team recommends very tight sizing. “Some of the RNA-seq methodologies, when you’re doing de novo sequencing of transcriptomes and want to do assemblies, tend to perform better when the size distribution of the library inserts is very tight,” he says.
The reason this is helpful for splice detection is in adding another dimension of data in the alignment step to allow for the inference of structure between known nucleotides. “If you know where the left read is, and where the right read is, and if you know the size of the fragments, then you can infer based on known exons the entire pattern in between,” Levine says. “You can calculate the likelihood that the exon is included or not based on where pairs of reads are.” The same holds for de novo assembly in general, he notes. With very tight size distribution, “you have a much more constrained situation when you’re assembling,” he adds.
Levine also believes that the Pippin platform is well suited for planning ahead as sequencing technologies evolve to offer longer reads. “As we need longer inserts, we’re going to be limited in terms of what we can get off the SPRIworks machine,” he says. Pippin’s ability to work with longer ranges means that “it’s extremely useful both for the ability to do sample preparation now and for the ability to work with these future technologies.”
Pippin Platform Recommended for Nextera Mate Pair Size Selection
Illumina released new sample prep protocol guidelines for generating mate pair libraries with its Nextera kit, and we’re pleased to report that the Pippin platform is the recommended choice for automated size selection.
You can check out the Nextera Mate Pair Sample Preparation Guide here . (We’re under Size Selection in Chapter 3, beginning on page 40 of the Guide.)
Illumina says that using an extra size selection step offers “more stringent” sizing than AMPure alone and lets users make libraries with larger fragments and more precise distribution than a gel-free approach. While the company has validated a manual approach in addition to the Pippin platform, Illumina’s guidelines note that “in our experience running a standard agarose gel does not provide as robust and reproducible results as the Sage Pippin Prep.”
In the user document, Illumina recommends the Pippin Prep with the 0.75% cassette and “eluting fragments with a broad range of sizes, of 3 to 6 kb in width, increasing in width with increasing fragment length (e.g. 2–5 kb, 4–8 kb or 6–12 kb).”
For current Pippin users, we would like to add that you can also use the 0.75% agarose dye-free cassette (BLF7510) with the BluePippin for equivalent results.
Poster: Exome Sequencing with Ion Torrent and Pippin Prep
In a poster from Ion Torrent (Life Technologies) for 2012’s ASHG, scientists looked at exome sequencing by studying a familial trio on both the PGM™ and the Proton™ instruments. Size selection for both sequencers was performed on the Pippin Prep from Sage Science.
Using an enrichment process targeting protein-coding exons from various genetic databases (including GenCode, RefSeq, Ensembl, and others), the scientists report “an on-target read mapping rate of 80%.” The sequencing, which took about four hours on either instrument, generated more than 5 Gb of aligned sequence, with average depth greater than 50x.
The Life Technologies authors note that they used Pippin size selection for both instruments, selecting an average length of 200 bp for the Proton and 300 bp for the PGM. (Check out figure 3 of the poster to see the nice clean peak they generated with Pippin.) This step was followed by exome enrichment and amplification prior to sequencing. The results indicate that sequencing exomes on the Proton is better than five times more efficient than on the PGM, and the authors say that stat is expected to improve even more.
Overall, the authors say, this study demonstrates that “the combination of focused exome enrichment and Ion Torrent Systems-based sequencing and analysis provides an efficient, accurate, and rapid means to detect genetic variation in the well-annotated portion of the genome for state-of-the-art genetic disease research.”
Check out the poster here.