Decoding Wild Rice Genomes: A New Era for Climate-Resilient Agriculture
Asian rice, domesticated around 10,000 years ago, is a dietary staple for more than one third of the global population. Yet, intensive breeding for improved yield has reduced its genetic diversity – making modern varieties increasingly vulnerable to stressors like heat, drought, and salinity. In contrast, wild rice relatives have adapted to a wide range of environmental conditions providing untapped genetic material for rice crop improvement.
A landmark study, published in Nature Genetics on April 28, 2025, traced the evolutionary history of wild rice species, classified in the genus Oryza, defining their phylogenetic relationships and deciphering how their genomes have been shaped by 15 million years of evolutionary history. The research was led by Prof. Rod Wing, Director of the Arizona Genomics Institute (AGI), and his postdoctoral research associate, Alice Fornasiero, of the King Abdullah University of Science and Technology (KAUST) in Saudi Arabia, in collaboration with Wageningen University & Research in the Netherlands.
Thanks to the sequencing of both genomic DNA and RNA of 11 wild rice species (i.e. two diploids and nine tetraploids) using the PacBio Sequel II and IIe instruments, the team could successfully generate ultra-high quality genome assemblies and use accurate full-length transcripts for gene annotation. The analysis of this valuable and publicly available genomic resource allowed the construction of the Oryza pangenome – i.e. the genomic representation of the entire genus – and the description of a “core” portion remarkably stable and a “dispensable” portion mainly composed of transposable elements. Due to their ability to move and reshuffle the genome, these elements has played a crucial role in shaping the genetic diversity in these species.
The analysis of gene expression in Oryza coarctata, a species adapted to saline conditions of coastal regions, showed an unusual balance between its two subgenomes, with both contributing equally to gene activity. This equilibrium may help explain the resistance of O. coarctata to salty environments.
These findings build a genetic foundation that could help scientists breed more resilient rice varieties, or even domesticate wild species through approaches like neodomestication to generate climate-ready crops for a more sustainable agriculture.
Use of Sage technology at the Arizona Genomics Institute’s Service Center:
For the wild rice genome project described above, the genomes were sequenced in both CLR and CCS/HiFi modes. The WGS libraries were size selected with the BluePippin, using 0.75% agarose cassettes with marker U1 (for CLR libraries) or marker S1 (for CCS/HiFi libraries ).
For current projects at AGI, the team has switched from the BluePippin to the PippinHT for its size selection needs. AGI has been an early access test site for the PippinHT Range+T program mode (for improving accuracy of HMW size selection) and the soon-to-be-released PippinHT 0.75% agarose cassette internal standards (which increase sample capacity).
SageHLS sample prep for ultra-long Nanopore Sequencing at NextOmics/GrandOmics
NextOmics/GrandOmics is the largest third-generation sequencing service company in China. The NextOmics division focuses on animal, plant and bacterial genome sequencing, while GrandOmics focuses on human genome sequencing.
GrandOmics became the first certified PromethIon service provider in China in June, 2018. In April, 2019, GrandOmics and Oxford Nanopore announced a strategic collaboration to sequence 100,000 Chinese genomes on the PromethIon platform by the end of 2021.
Recently, NextOmics/GrandOmics has disclosed some examples of its PromethIon output on Twitter (see below), and shared with Sage Science an overview of their whole genome workflow.
1) Input material – human cultured cell lines, young plant leaves.
2) Extraction is carried out using a modified phenol/chloroform method.
3) For ultra-long reads, size selection is performed using the SageHLS instrument.
4) SageHLS input: not more than 10ug per lane.
5) SageHLS size selection program(options, depending on extraction quality): High-Pass 50kb, 100kb, 250kb, 300kb, 350kb, 500kb etc. (collection stage only)
6) Oxford library kit: SQK-LSK109 Ligation kit.
Typical size distributions from the PromethIon flow cell show read N50 values as high as 140kb, with an impressive 28% of the reads longer than 200kb.
[click the images to enlarge]
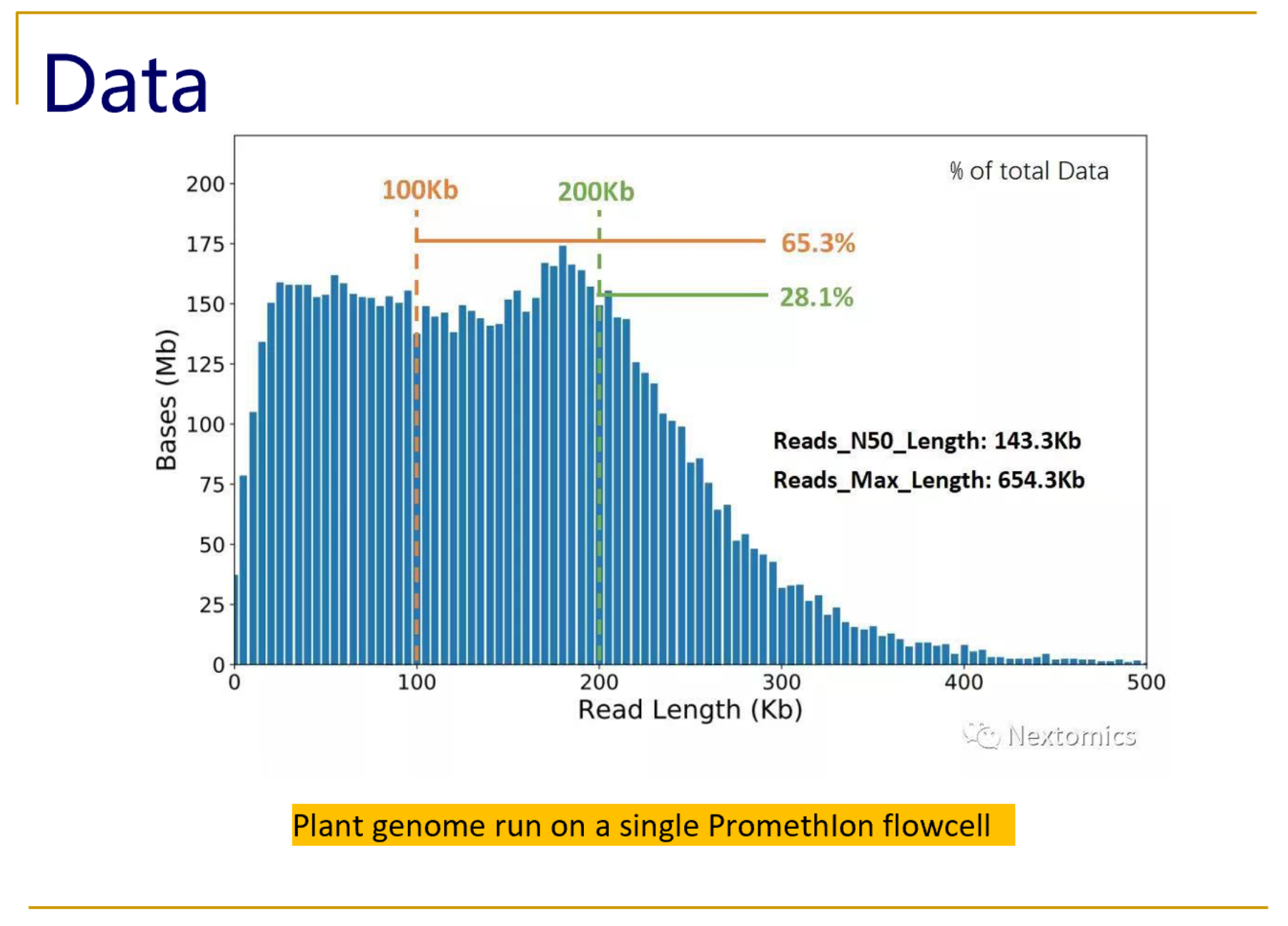
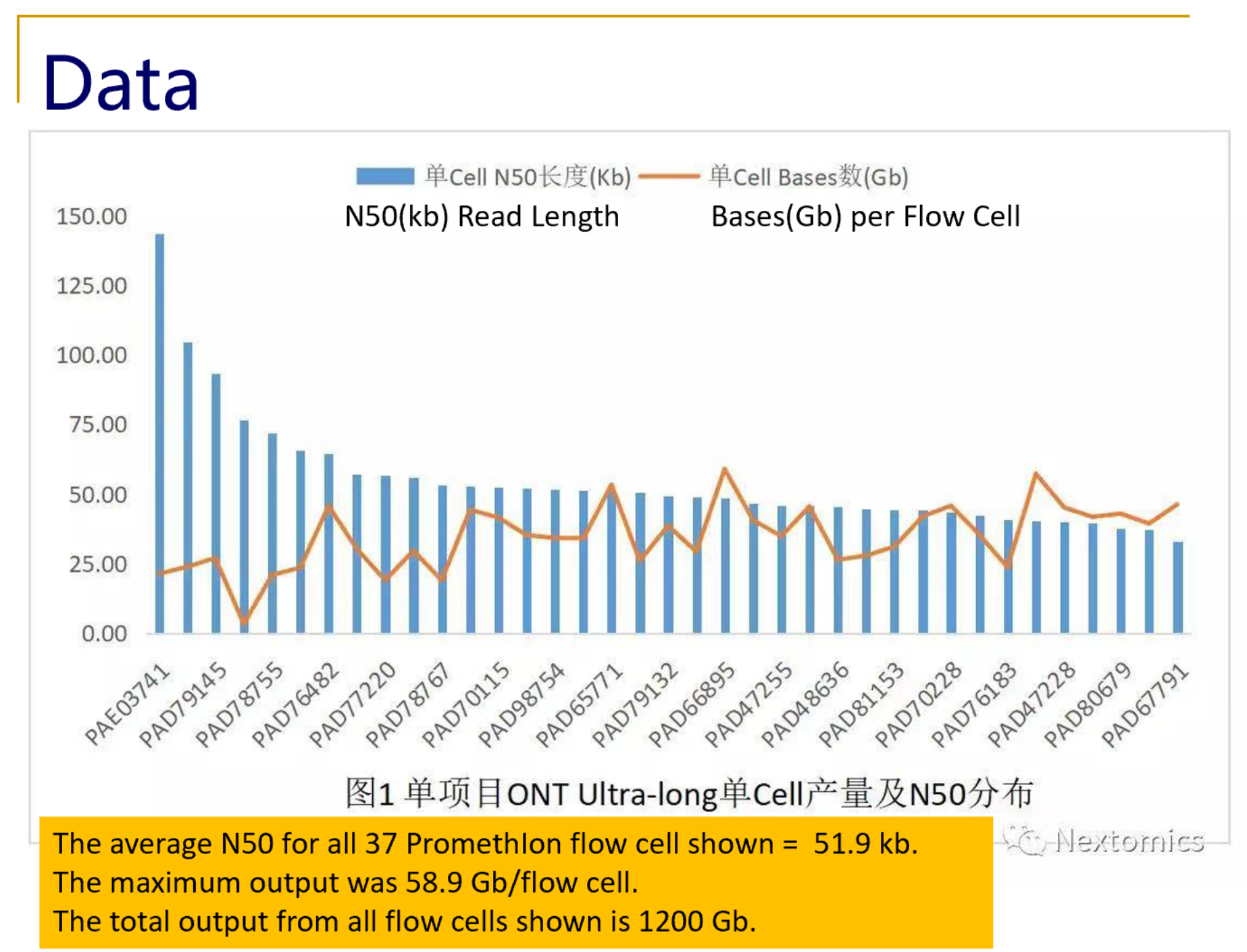
What NextOmics/GrandOmics says about SageHLS for ultra-long read PromethIon sequencing:
“The bottleneck of ONT sequencing is sample preparation, especially for ultra-long sequencing. The ONT user should optimize the extraction steps in order to protect the DNA from shear forces generated during liquid handling. After the extraction, we use SageHLS size-selection to remove shorter DNA fragments and enrich the ultra-high molecular weight DNA. SageHLS is a key component of our ultra-long PromethIon workflow.”
**We would like to express our gratitude to our great distributor, APG Bio, for their help with this post!
Tracking Yellow Fever: New Study Aims To Tackle YFV Epidemics
Last year, we posted a story about an international team of scientists who embarked on a mission with Brazilian researchers to study the dangerous mosquito-borne Chikungunya virus (“Tracking Chikungunya: New Study Traces Outbreak Path”- we were pleased to donate a Pippin Prep to the effort.)
Recently the team published a new study in Science, having deployed NGS to elucidate the epidemiology of the Yellow Fever virus in Brazil: “Genomic and epidemiological monitoring of yellow fever virus transmission potential”. According to the authors, virological surveillance requirements are to “ (i) track epidemic origins and transmission hotspots, (ii) characterize genetic diversity to aid molecular diagnostics, (iii) detect viral mutations associated with disease severity, and (iv) exclude the possibility that human cases are caused by vaccine reversion.” By sampling humans and non-human primates (NHP) across the state of Minas Gerais, the epicenter of a 2017 outbreak, the researchers analyzed how the virus spreads through space, between humans and NHPs, and the “contribution of the urban cycle”. The authors note that this type of real-time monitoring can contribute to global efforts to eliminate future epidemics – and one would assume, potentially save lives.
Our minor contribution to this work was based on outreach from Antonio Charlys Da-Costa, who had used a Pippin Prep as part of his studies at Dr. Eric Delwart’s lab at the UCSF Blood Systems Research Institute. Dr. Da-Costa, now at the Sao Paulo Institute of Tropical Medicine, was able to marshal resources from a number of other suppliers for this cause, including Illumina, Zymo Research, and Promega (surely there were others, apologies for the non-mention). Congratulations to the whole team, it’s inspiring to see such diverse institutions and agencies pulling these large-scale efforts together – and individuals like Antonio who went the extra mile.
An interesting side note, a number of other important viral genomic findings were also published during this time by a consortium of Brazilan labs in collaboration with Dr. Delwart and UCSF:
Wuhan large pig roundworm virus identified in human feces in Brazil
At Mount Sinai, Precise Sizing and SMRT Sequencing Yield Unprecedented Read Length
At the Icahn Institute for Genomics and Multiscale Biology at Mount Sinai in New York City, scientists use automated DNA sizing together with long-read sequencing to analyze clinical samples, conduct routine surveillance on microbes, and more.
Technology development expert Robert Sebra, Ph.D., relies on Single Molecule, Real-Time (SMRT®) Sequencing from Pacific Biosciences with BluePippin™ automated DNA size selection from Sage Science. Together, these tools offer a powerful solution and industry-leading read lengths that allow Sebra and other researchers to resolve repeat elements and structural variants, rapidly close microbial genomes, and measure epigenetic marks.
Sebra, an assistant professor of genetic and genomic sciences, is no stranger to the SMRT Sequencing platform: he spent five years working at PacBio helping to develop that technology. Ultimately, his belief in the system led him to join the Icahn Institute, where he would get to use the PacBio® sequencer in the field. “There was a lot to be gained by taking the technology and applying it in a clinical setting,” says Sebra, who came to Mount Sinai in 2012. “I had experienced firsthand the value of long-read sequencing and wanted to apply it to human and infectious disease research.”
Since its founding by Eric Schadt in 2011, the Icahn Institute has attracted some 150 leading scientists and clinicians who bring a network-based approach to various biological questions, many of them focused on cancer, Alzheimer’s disease, allergy and asthma, and infectious disease. Among the institute’s well-stocked core facilities are two PacBio RS II sequencers and a BluePippin instrument, which are used together for projects requiring extra-long reads.
The PacBio RS II is his go-to system for epigenetic profiling, finishing microbial genomes, and exploring DNA samples likely to have repeats, large structural rearrangements, or ones that require allelic or accessory genome phasing.
As he applies long-read sequencing to these projects, Sebra continually looks for ways to generate the longest possible reads. One complementary technology for the PacBio workflow is BluePippin, an automated DNA size selection platform from Sage Science. Removing smaller fragments from the sequencing library ensures that the PacBio platform focuses on the longest fragments, so accurate sizing can improve average read length considerably. “You could do a traditional pulsed field gel every time you’re trying to size select, but it takes too much time, doesn’t scale well, and the DNA input requirement is really high,” Sebra says. “BluePippin is fast and cheap, and it’s the only option for size selecting in a high-throughput fashion. We purchased one as soon as it was available.”
Since bringing in BluePippin in 2012, Sebra’s team has run more than 100 libraries using the BluePippin+PacBio combo — in fact, he says, “For projects requiring near finished genome assembly, I don’t think we’ve prepared a library without BluePippin size select since owning the instrument.” He has been pleased with the amount of size-selected library the technology yields, noting that in virtually every experiment it produces more than enough to sequence a genome to completion on the PacBio RS II. He generally excludes all fragments smaller than 10 Kb to target the ultra long fragments, but says that in cases where input DNA is especially low or the genome is quite large and requires more library, he lowers that threshold to 7 Kb.
Sebra notes that the size selection step has exceeded his expectations for overall improvement in read length and throughput of SMRT Sequencing. The boost to mean read length from adding BluePippin size selection ranges from about 30 percent to 125 percent, depending on the input quality, he says.
In one infectious disease study, the team sequenced multiple MRSA isolates using PacBio with and without BluePippin sizing, finding that prior to sizing, 50 percent of the bases are in reads 5 Kb or longer, while after sizing that number more than doubled to 12.5 Kb.
“If your throughput of [PacBio] runs is high enough, a BluePippin is really pretty affordable,” Sebra says. “Size selection reduces the number of SMRT Cells required to achieve a particular sequencing goal, so it pays for itself pretty quickly.”
For more about Sebra’s scientific and clinical efforts, check out the full case study here.