At the University of Florida, SageELF Delivers Better Sizing for ddRAD-seq and Long-Read Pipelines
 Clients of the NextGen DNA Sequencing core at the University of Florida in Gainesville rely on Scientific Director David Moraga Amador to find and validate the best technologies for their projects. In addition to bringing in the best sequencers, that means hunting for sample prep methods and instruments that make downstream results more reliable and reproducible.
Clients of the NextGen DNA Sequencing core at the University of Florida in Gainesville rely on Scientific Director David Moraga Amador to find and validate the best technologies for their projects. In addition to bringing in the best sequencers, that means hunting for sample prep methods and instruments that make downstream results more reliable and reproducible.
For scientists using a custom ddRAD-seq protocol with Illumina sequencing, the core lab team recommended SageELF, which performs whole-sample fractionation and splits input DNA by size into 12 contiguous fractions. These clients had been coming to the lab with libraries that were challenging to sequence cleanly because of their wide size distribution. “These libraries often look very ugly: they have a broad range size distribution, multiple peaks, and they’re very difficult to quantitate,” Moraga Amador says. “Fragments might go from hundreds of base pairs to ten thousand base pairs.” To address the problem, the team began running these libraries on the SageELF and delivering fractions back to users with a TapeStation analysis of fragment size; the clients then choose which to advance to sequencing. “Our users love the fact that these peaks are so sharp, the sequencing output is predictable, and the quality metrics are improved,” he says. “Now we have five or six groups doing this routinely.”
Years before that, Moraga Amador had become a Sage Science customer when he introduced the PacBio sequencing platform to his core lab and brought in a BluePippin to increase average read lengths. When the SageELF launched more recently, he saw an opportunity to maintain the precise size selection he was used to while making more of each sample.
With BluePippin, Moraga Amador and his team used the high-pass protocol for long-read sequencing (PacBio RS II), collecting all fragments longer than a certain size. The smaller fragments were tossed out as part of the size selection process. With SageELF, he can use the whole sample, allocating each fraction to the part of the project where it will have the most value. “All of those fractions are good,” Moraga Amador says. “They turn into very sharp fractions and they sequence beautifully on the instrument.” SageELF lets his team size DNA up to 30 Kb, fitting nicely with the requirements for the PacBio instrument.
Moraga Amador says the device is also useful for PacBio Iso-Seq projects, where protocols require binning DNA by size prior to sequencing. “With the SageELF we just do one single run and collect all the fragments for our samples, and then pool the fragments of like size,” he says. “The key for us is doing a single run so we don’t waste any of the sample.” SageELF allows his team to generate the needed fractions from much less sample DNA than if they had to size each fraction individually.
The SageELF system was easy to set up and start running, Moraga Amador notes. “It didn’t take a lot of practice,” he says, noting that the team set up the instrument themselves with a little phone support from Sage Science. For most projects, there’s no tinkering required to get the results they expect. “There is very little optimization we have to do if we follow the instrument recommendations,” he adds.
Poster: Better Cell-Free DNA Analysis with Pippin + Rubicon Genomics
Recently we blogged about the rise of cell-free DNA studies, and how precise size selection is one tool that can help researchers isolate DNA of interest for further analysis (such as sorting out fetal from maternal genetic material).
At the AGBT Precision Health meeting in Scottsdale, Ariz., last month, we teamed up with Rubicon Genomics to present a poster demonstrating how our technologies can be used together to generate better results from cell-free DNA studies. (If you were at the meeting, it was poster #107.)
For this work, we performed pre-library size selection using the Pippin Prep to separate fragments 160 bp and larger (since that’s about the size of the mononucleosomal unit) from smaller fragments, which are typically the ones scientists want to analyze for cancer or prenatal studies. After sizing, we prepared libraries for sequencing using Rubicon Genomics’ ThruPLEX® Plasma-Seq kit, which is designed for low-input samples from plasma and liquid biopsies like cfDNA. Libraries were also prepared and analyzed without size selection so we could see how much of a difference that step made.
Even though we started with less than 1 ng of size-selected material, we were able to produce highly diverse libraries that were significantly enriched for cell-free DNA fragments between 60 bp and 120 bp. That enrichment did not happen for libraries prepared without size selection. This graph of insert size distribution shows how clearly delineated the shorter fragments were when size selection was used.
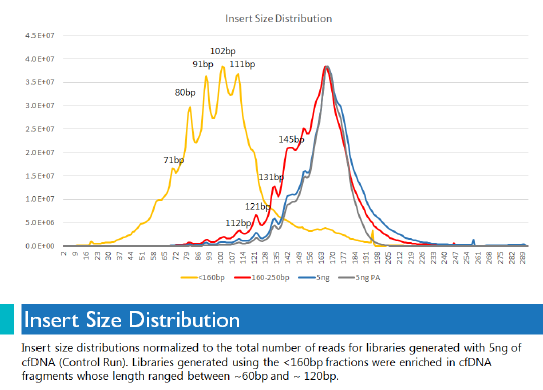
While Pippin Prep was used for this project, Sage customers can use PippinHT as well to perform what we call “low-pass” sizing — that is, removing everything larger than a certain size. We look forward to seeing how our users deploy automated size selection technology to enhance their cell-free DNA studies.
Size Selection Has Utility for Cell-Free DNA Studies
The rise of research studies and diagnostic tests looking at cell-free DNA — particularly fetal DNA in a mother’s bloodstream — has happened with astonishing speed. Prenatal genetic testing, for instance, has already supplanted many invasive clinical tests such as amniocentesis or chorionic villus sampling. Cell-free DNA is now considered an important source of information about cancer, and will no doubt have many other applications as we learn more about it.
These studies are particularly interesting to us because isolating cell-free DNA involves accurate size selection. Foundational research has consistently found that cell-free fetal DNA is shorter than cell-free maternal DNA: this early study determined that fetal DNA was less than 300 bp, while maternal DNA was larger than 1 Kb, while another study reported a dominant peak of about 160 bp for fetal DNA.
A more recent publication explored various methods of analyzing fragment sizes for a study of cell-free fetal DNA. With paired-end sequencing as well as basic electrophoresis (sizes were read with a Bioanalyzer), the scientists were able to distinguish maternal from fetal DNA. With extremely specific findings of fragment size, they were also able to detect some cases of fetal chromosomal aneuploidy just by observing size aberrations.
We’re excited about the possibilities of applying automated DNA size selection to cell-free DNA studies. Other methods of size selection have not been terribly successful due to the yield challenge; DNA derived from a fetus or tumor is already such a small proportion of DNA in these samples. But with a platform like ours, which significantly boosts yield compared to other sizing techniques, we think there is great potential for enhancing cell-free DNA research.
We’re presenting a poster on this topic at the AGBT Precision Health meeting right now. If you’re attending the conference, check out poster #107 — and if not, we’ll have more details on our blog next week.
Where Illumina Users Gather, Great Science Is on Display
The Sage team is pleased to be sponsoring the slate of upcoming Illumina user group meetings. We’ve attended many of these events over the years, and they’re excellent venues that showcase truly impressive work from the company’s broad customer base. We learn something new at each meeting we attend!
Sage instruments are important for a number of applications related to Illumina sequencing, from Nextera library preparation and PCR-free libraries to paired-end and mate-pair libraries. For more details on specific applications, check out this resource page.
For a good sense of how Illumina users are applying our Pippin family of automated DNA size selection platforms, don’t miss our frequently updated list of citations from peer-reviewed literature. And if you’ll be at the user group meetings or other upcoming genomics conferences, please stop by the Sage booth! We’d love to meet you and learn more about your research.
At the University of Delaware, Early Adopter Genomics Core Spots New Science Trends
 The genomics core facility at the University of Delaware has set itself apart from other service providers by being among the first to adopt new sequencing technologies. The strategy has been a success: today, the facility serves customers around the world, hailing from research and nonprofit institutes, federal agencies, and even foreign governments. While projects range from microbial to human and everything in between, agrigenomic studies are especially popular for users looking to improve growth and disease resistance among crops and livestock.
The genomics core facility at the University of Delaware has set itself apart from other service providers by being among the first to adopt new sequencing technologies. The strategy has been a success: today, the facility serves customers around the world, hailing from research and nonprofit institutes, federal agencies, and even foreign governments. While projects range from microbial to human and everything in between, agrigenomic studies are especially popular for users looking to improve growth and disease resistance among crops and livestock.
Bruce Kingham, who runs the genomics core lab, has also focused on adopting state-of-the-art tools to keep the sequencers running happily. Size selection has been essential for delivering optimal results to his user base, from their first Illumina NGS platform in 2007 to the PacBio single-molecule sequencing system. It was the acquisition of the Illumina GA that spurred his team to offer library prep as a service, for which they invested in the Pippin Prep for automated DNA size selection. “That allowed us to not only get a very focused size for the libraries that we were preparing, but more importantly it allowed us to start with a much smaller quantity of DNA,” he says. Prior techniques relied on inefficient fragmentation procedures and gel extraction to isolate the desired fragment size, resulting in a great deal of undesirable sample loss.
Today, Pippin sizing — now with BluePippin — continues to be important for Kingham’s Illumina workflow, including PCR-free projects. “Size selection has been critical because the PCR-free library preparation process can be prone to generating libraries that have a broader size range,” he says. “Illumina technology for a number of reasons does not like libraries that are broad in size.” From clustering efficiency to optical analysis, these sequencers perform best when fed libraries with tightly sized DNA fragments. For Illumina sequencing in general, Kingham says, “downstream analysis, including mapping or de novo assembly, is going to be more efficient and have more statistical significance if the size range of individual libraries is focused.”
For PacBio sequencing, Kingham’s team uses both BluePippin and SageELF for size selection. Because the BluePippin is so useful for eliminating small fragments and keeping the PacBio platform focused on generating the longest reads possible, it dramatically improves the quality of results. “With the volume of sequencing that we do, the BluePippin paid for itself in a couple of months,” Kingham says. By increasing average read length and N50 read length, BluePippin “lowers the cost of the data that needs to be generated to achieve a certain sequencing goal, such as the lowest number of contigs,” he adds. The lab uses SageELF for Iso-Seq protocols, where it significantly reduces the amount of input DNA required.
Looking ahead, Kingham sees increased demand from scientists for pairing genomics and proteomics data. It’s a trend that fits nicely at his home institute, which has a mission of promoting interdisciplinary research. To that end, his team has already begun evaluating the SageELF for use in protein fractionation. “That could be a welcome service, and I’m always looking for new services to provide,” Kingham says. “I want to see my instruments running as much as possible.”