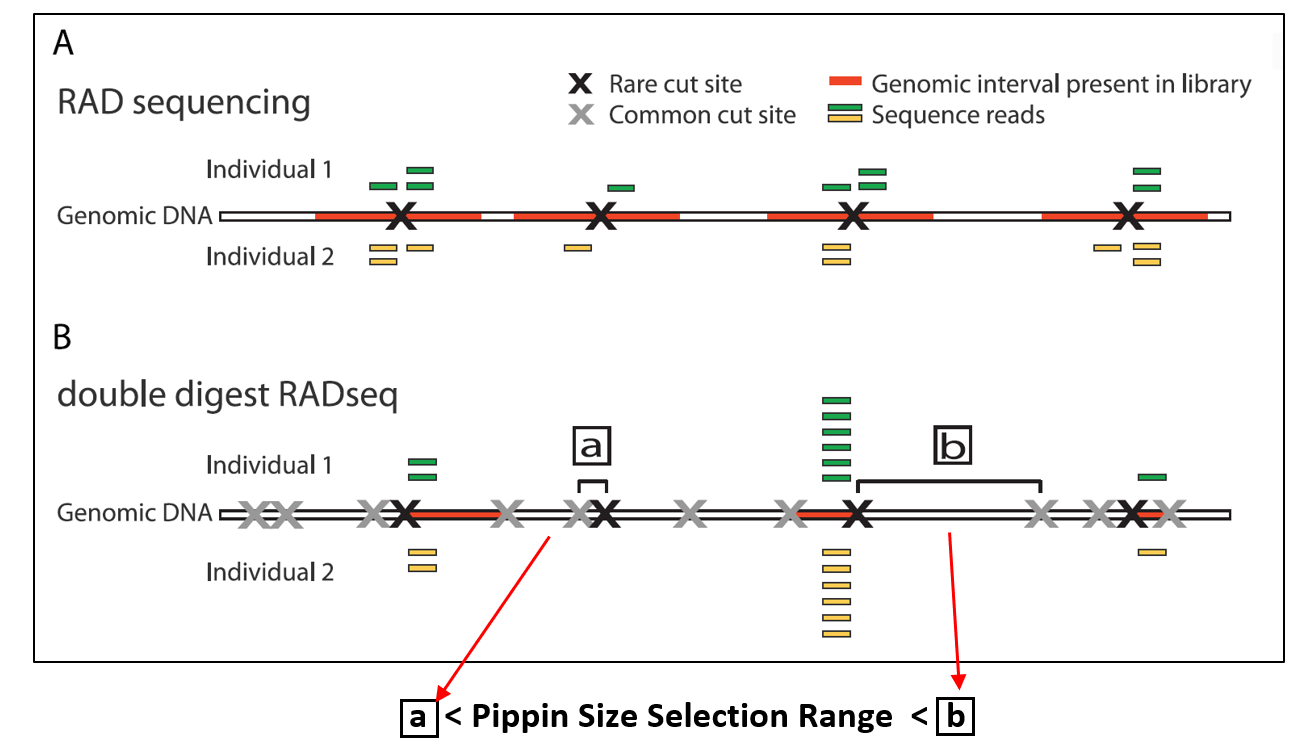
An improvement on the RAD-seq genotyping method (2011, Baird et al. PLOS One). With the RAD-seq method, individual genomes are sheared and digested using a restriction enzyme. Barcoded primers that tag the individuals are ligated to the restriction sites and sequenced. The identification of SNPs are used to genotype the individuals for population or QTL studies. This is a cost-effective and powerful tool for comparing individuals using de novo reduced representation sequencing.
The ddRAD-seq is a method that adds a second restriction enzyme and uses precise Pippin size selection to improve RAD-seq genotyping. By combining a second more common cut site to the RAD-seq method, genomic intervals that are larger and smaller than the size selection range are eliminated. This helps normalize the comparative sequences, improves the accuracy of the genotyping, and thus reduces cost. Since its emergence in 2011, an number of variations of the RAD-seq method have been developed, many of which incorporate Pippin Size Selection (e.g. EpiRad-seq (epigenetic data), HyRAD-seq (degraded samples), and SLAF-seq (large populations)).
Links:
Foundational paper:
(Peterson et al 2012) Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species
Recent Study:
(Smith et al 2022) Signatures of selection underpinning rapid coral adaptation to the world’s warmest reefs